- Software
- Open access
- Published:
Gene Expression Browser: large-scale and cross-experiment microarray data integration, management, search & visualization
BMC Bioinformatics volume 11, Article number: 433 (2010)
Abstract
Background
In the last decade, a large amount of microarray gene expression data has been accumulated in public repositories. Integrating and analyzing high-throughput gene expression data have become key activities for exploring gene functions, gene networks and biological pathways. Effectively utilizing these invaluable microarray data remains challenging due to a lack of powerful tools to integrate large-scale gene-expression information across diverse experiments and to search and visualize a large number of gene-expression data points.
Results
Gene Expression Browser is a microarray data integration, management and processing system with web-based search and visualization functions. An innovative method has been developed to define a treatment over a control for every microarray experiment to standardize and make microarray data from different experiments homogeneous. In the browser, data are pre-processed offline and the resulting data points are visualized online with a 2-layer dynamic web display. Users can view all treatments over control that affect the expression of a selected gene via Gene View, and view all genes that change in a selected treatment over control via treatment over control View. Users can also check the changes of expression profiles of a set of either the treatments over control or genes via Slide View. In addition, the relationships between genes and treatments over control are computed according to gene expression ratio and are shown as co-responsive genes and co-regulation treatments over control.
Conclusion
Gene Expression Browser is composed of a set of software tools, including a data extraction tool, a microarray data-management system, a data-annotation tool, a microarray data-processing pipeline, and a data search & visualization tool. The browser is deployed as a free public web service (http://www.ExpressionBrowser.com) that integrates 301 ATH1 gene microarray experiments from public data repositories (viz. the Gene Expression Omnibus repository at the National Center for Biotechnology Information and Nottingham Arabidopsis Stock Center). The set of Gene Expression Browser software tools can be easily applied to the large-scale expression data generated by other platforms and in other species.
Background
A microarray measures the expression of thousands of genes simultaneously. This experimental system has revolutionized biological research by enabling discovery of a large set of genes whose expression levels reflect a given cell type, treatment, disease or development stage. Since the advent of this technology more than a decade ago, a large amount of expression data has been accumulated on more than 100 species [1]. Several initiatives have been undertaken to develop microarray public data repositories and analysis tools for scientists to share and utilize these data [2]. The public data repositories, such as NASC, NCBI GEO [3], EBI ArrayExpress [4, 5] and NIG CIBEX [6], have been collecting, annotating, storing and redistributing large amounts of microarray data from diverse experiments. For example, NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) has collected 366,965 samples from 14,304 experiments. These microarray data are invaluable resources for scientific research and discovery.
Effective utilization of these datasets has, however, been limited because of a shortage of suitable tools to integrate large-scale and diverse microarray datasets. In most common use case, a scientist performs an experiment-based analysis: he or she downloads microarray data and sample annotations corresponding to a single experiment, inputs the data into a microarray data-analysis tool, such as GeneSpring [2], HDBStat! [7], or Bioconductor packages [2], etc., and carries out single-experiment centered analysis. In another common use case (e.g. for many gene-centric studies), a scientist wants to know how the expression of a given gene changes under various experimental conditions. The latter case is critically important for discovering gene functions, validating biomarkers, and developing new drugs targeted to specific genes. To answer gene-centric questions, we must have a tool that can be used to integrate a large amount of data from different microarray experiments. Developing such a tool presents several challenges.
The first challenge is the heterogeneity of data collected from different microarray experiments. Different microarray experiments from different laboratories are usually designed independently for specific research purposes. Heterogeneity might come from differences in experimental designs, materials sampled, developmental stages, treatment levels (including controls), and so on. The second challenge is to develop an effective software tool to process such a large amount of data at an acceptable speed with currently available hardware resources (i.e., CPU, memory and network). The third challenge is related to the complexity of displaying or visualizing data in a software tool. Most software tools, when applied to large data sets, display items in an extended page or multiple display pages. Therefore, it is impossible for users to get an overall view of the data on a single page. It is also inefficient and inconvenient for users to scroll display pages to find interesting information from thousands of data items. Thus, it is important to design a data display interface that can show both an overall view of a large-scale dataset in its totality and a detailed view of individual data points.
Genevestigator [8] and GeneChaser [1] are two web-based gene expression visualization tools that have successfully integrated a large number of microarray datasets and facilitated gene-centric and cross-experiment gene-expression discoveries. Genevestigator defines experiment annotation categories as Tissues/Organs, Developmental Stage, Environmental Factors (Stimulus) and Mutation. The expression data and the analysis results are organized according to these categories. The microarray experiments are discarded if they cannot be classified into one of the predefined categories. GeneChaser, on the other hand, automatically re-annotates and analyzes GDS datasets from NCBI GEO. It segregates all experimental conditions (treatment levels) into groups and then performs group versus group comparisons. However, the display systems of both Genevestigator and GeneChaser are limited. These two tools display data with heatmap or bar graphics on a display page with extended dimension or in multiple display pages. Only a limited number of data points can be shown at a time. Users have to scroll down the page to find interesting data points from among hundreds or thousands of total experimental conditions.
The GEB, on the other hand, displays efficiently a large number of data points simultaneously. This has been achieved by developing a set of software tools of data extraction, data management, data annotation, data processing, and gene expression profile search & visualization. This set of software tools can be applied to microarray data in both public and private data repositories. The current public GEB web service (http://www.ExpressionBrowser.com) integrates 301 ATH1 microarray experiments that were originally stored in the data repositories of NCBI and NASC [9]. Arabidopsis, as a model plant, is widely used in various microarray experiments and gene-network modeling [10–12]. The results and knowledge obtained from Arabidopsis studies can be used as a reference for corresponding research on other plants, especially field crops [13, 14].
Implementation
Overall design of workflow
The GEB workflow is shown in Figure 1. Microarray data can be downloaded from public data repositories with the data extraction tool. Alternatively, data owners may upload their data directly into GEB. The data extraction tool harvests raw data files, sample annotations, and experimental designs from data repositories into the GEB data-management system. Data curators use the web-based interfaces of the data-management system to create sample sets by combing all replicated samples in each treatment level into individual groups (i.e. sample sets). Then, the data curators define a T/C by selecting a treatment sample set and a control sample set. In the data-processing pipeline, the microarray data are normalized, and the log2 ratio of treatment-over-control (LOG2R) and its t-test P value are calculated. The normalized intensities of each chip, average intensities of each sample set, LOG2Rs and P values of each T/C are loaded into the GEB database, from which the data can be queried via the web-based search & visualization tool.

The schema and workflow of GEB. Microarray data can be downloaded into GEB from public repositories or uploaded into GEB by data owners. GEB is composed of a set of functional components. The major components are the data extractor (a command-line program), the data management system (a web application), the data processing pipeline (a set of command-line programs piped together), a MySQL database, and a web-based search and visualization tool.
Affymetrix probe set annotation
The probe sets on Affymetrix ATH1 chip were annotated via the following procedures: (1) Arabidopsis cDNA sequences and annotations were downloaded from TAIR (http://www.arabidopsis.org/) and ATH1 probe sequences were downloaded from Affymetrix; (2) All probe sequences were BLASTed against all cDNA sequences; (3) A probe set was mapped to a cDNA when nine or more probes in the probe set had a 100% match to a cDNA sequence (each ATH1 probe set contains 11 probes); and (4) The annotation of matched cDNA was used as the annotation of the probe set.
Data extraction and management
The data extraction tool was developed using Java with Jakarta Commons Net Library (http://commons.apache.org/net/). The tool is a web crawler that recursively harvests raw data (such as Affymetrix CEL files), sample annotations, and experiment design descriptions from a repository website and then loads them into GEB database. To download data from different repositories, a corresponding plug-in component was developed for each repository. So far, two data extraction plug-ins have been developed for harvesting data from GEO and NASC.
The data-management system was developed for data curators to view and annotate the microarray data extracted from data repositories or submitted by data owners. Data curators annotate the data via the following steps:
First, a data curator creates a sample set by grouping replicated samples from every treatment level. The user interface for defining a sample set is shown in Figure 2A. A sample set name of "Wildtype_no treatment" is given at Name box and two replicates of "Wildtype_no treatment_Rep1" and "Widetype_no treatment_Rep2" are assigned to the sample set by moving them from the left panel to the right panel. Other sample sets in the experiment are created via the same procedure as noted above.

Screenshots of the GEB data management system. A. The web interface used by a data curator to define sample sets for all experiment data. B. The web interface used by a data curator to choose a treatment sample set and a control sample set to create a T/C. Detailed information for all eight sample sets can be found at http://expressionbrowser.com/arab/displayExperiment.jsp?id=2202517&tab=2.
Second, a data curator creates a T/C pair by choosing a treatment sample set and the corresponding control sample set from a drop-down menu (Figure 2B). For instance, we selected "ice1_no treatment" as treatment and "Wildtype_no treatment" as control to form a T/C. Then, the curator specifies a name of "ICE1 mutant vs. wild type" at Name box and detailed T/C information is given in Description box at the lower panel of Figure 2B. The control sample set is selected for a given treatment sample set so that only one-factor differs between the treatment and the control. Therefore, the biological effect of the T/C will be clearly distinguished by the differential factor. All possible T/C pairs were created in this way. In the example shown in Figure 2, a total of 10 T/Cs are defined as follows: 3 T/Cs for cold effects in a mutant (viz. "Ice1 mutant with cold treatment for 3 hr vs. Ice1 mutant with no treatment", "Ice1 mutant with cold treatment for 6 hr vs. Ice1 mutant with no treatment", and "Ice1 mutant with cold treatment for 24 hr vs. Ice1 mutant with no treatment"); 3 T/Cs for cold effects in wild type (viz. "Wildtype with cold treatment for 3 hr vs. Wildtype with no treatment", "Wildtype with cold treatment for 6 hr vs. Wildtype with no treatment", and "Wildtype with cold treatment for 24 hr vs. Wildtype with no treatment"); 3 T/Cs for mutation effects under cold treatment (viz. "Ice1 mutant with cold treatment for 3 hr vs. Wildtype with cold treatment for 3 hr", "Ice1 mutant with cold treatment for 6 hr vs. Wildtype with cold treatment for 6 hr", and "Ice1 mutant with cold treatment for 24 hr vs. Wildtype with cold treatment for 24 hr"); and one T/C for mutation effects without cold treatment (viz. "Ice1 mutant with no treatment vs. Wildtype with no treatment"). All 10 T/Cs are shown at http://expressionbrowser.com/arab/displayExperiment.jsp?id=2202517&tab=1. After all treatment levels in each experiment are transformed into T/Cs, different experiments have same data structure and are comparable to one another and are, thus, easily integrated together. As a result, the heterogeneity caused by the differences in experimental designs is removed. The LOG2R of T/C also removes system errors that affect both treatment and control. Therefore, the ratio data generated based on T/Cs can be more instructive and reliable than intensity data generated from treatment levels.
Data processing and data quality monitoring
The GEB data-processing pipeline is composed of four consecutive programs. The first program is for data normalization using the Robust Multichip Average (RMA) algorithm [15] that was implemented in the Bioconductor Affy package (http://www.bioconductor.org/packages/2.4/bioc/html/affy.html). The second program takes this normalized intensity data as input and computes average intensities, standard deviations, LOG2Rs, and P values of two-sample, two-tailed t-tests. The third program renders JPEG images of MA plots [16, 17] with average intensity as the x-axis, LOG2R as the y-axis, and P value as the color. The images are loaded into the GEB application server (Tomcat) for data display when queried by users. The fourth program computes the mean percentage coefficient of variation (%CV) of all microarray features (genes) in a sample set using the following two steps. First, the standard deviation, mean, and %CV of each feature (gene) in a sample set are calculated: that is, %CV = 100 * (Mean intensity/Standard deviation). Second, the mean %CV of all features in the sample set is calculated. The mean %CV of each of individual sample set is computed via the above procedure; the distribution of all mean %CVs is shown in Figure 3. Most sample sets have mean %CV between 0.5 and 4.68. There is a long tail to the right side of the distribution, in which the mean %CV ranges from 4.68 to 16. This result indicates that about 10% of the total sample sets have extremely large mean %CV, and thus probably have poor data quality. Mean %CV of a sample set could be used to monitor quality of the sample set because higher mean %CV implies larger variation among the replicated samples in the sample set. Therefore, any finding or conclusion from a sample set with high mean %CV must be interpreted cautiously. We plan to filter out the sample sets with extremely high mean %CV in the future to guarantee the quality of all the data in GEB.

The distribution of mean %CV of all sample sets. The mean %CV is calculated in two steps: first calculate the standard deviation, mean, and %CV of each gene in a sample set (%CV = 100 * Mean intensity/Standard deviation), and then compute the mean %CV of all genes in the sample set.
Some microarray experiments in NASC or GEO were discarded because there were no replicated samples or no suitable controls. As of now, there are a total of 301 experiments, 1450 T/Cs, and 33,074,500 LOG2R data points in the Arabidopsis GEB database. Additional data, when available, can be easily entered into GEB.
Data search and visualization
The Lucene search engine (http://lucene.apache.org/) is used for full-text search. Search index files in GEB are built with the text from gene identifiers, gene symbols, gene annotations, T/C names, T/C descriptions, experiment titles, and experiment descriptions. Genes, T/Cs, and experiments are searchable by matching keywords in the index files.
A 2-layer visualization display is designed to show large-scale data points as both an overall view and a detailed view. This visualization was developed using AJAX technology [18]. The first display layer is a static display (image) generated offline that contains all data points. The second layer is a real-time interactive display built by Web2.0 technology (JavaScript/AJAX). With the 2-layer display, users not only obtain an overall expression profile of the distribution of data points on the static plot, but can also get detailed information on each data point by real-time interactive searching or highlighting. The P value of ratio data is shown by the color of the data. Therefore, data significance level is displayed at the same time as the magnitude of the data is.
Results and Discussion
Full-text search
With full-text searching, users can easily access the information inside GEB. The full text searching method employed by GEB is different from the searching in Genevestigator [8] or GeneChaser [1], in which only gene identifiers or symbols can be used for searching. Users can obtain expression information from Genevestigator or GeneChaser only when they clearly know the gene names or symbols. In contrast, GEB carries out full-text search for any word or letters for a gene symbol, gene annotation, T/C name, T/C description, experiment title and experiment description. Users can freely explore the expression data with any search term they wish.
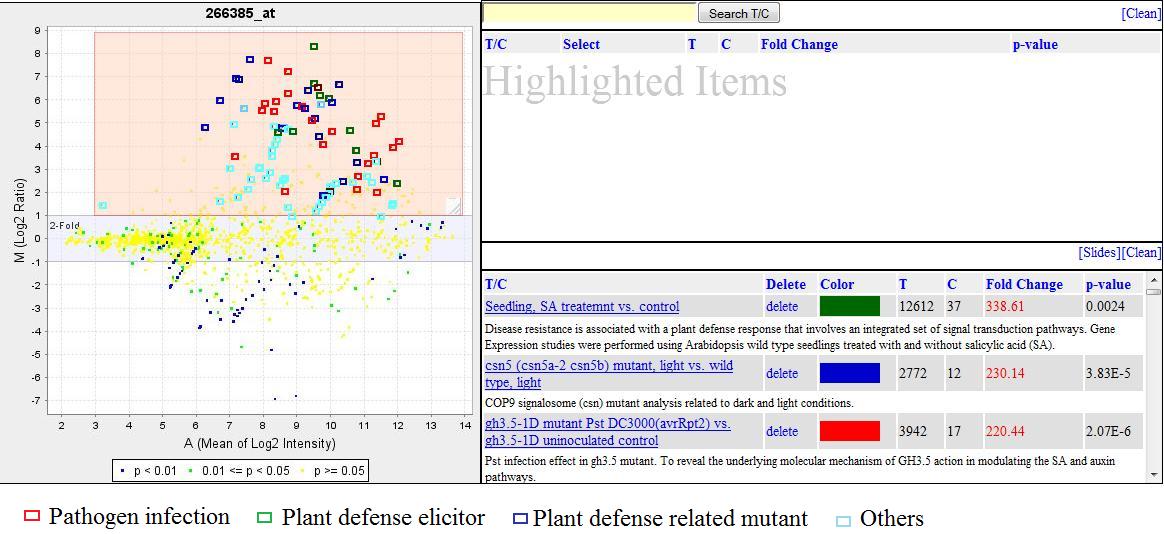
The full-text search is implemented in three places. The first is the GEB home page (http://www.ExpressionBrowser.com), where the user can enter keywords and find three types of information: genes, T/Cs and experiments. The second place is in Gene View (Figure 4), where users can search T/Cs and investigate how different T/Cs affect the expression of the selected gene. The third place is in the T/C View (Figure 5), where users can search genes and observe how the expressions of these genes are changed by the selected T/C.

The Gene View of PR-1 that is a disease-related gene. The up-regulation T/Cs were highlighted and selected. The color can be changed by right clicking on the color icon in the lower box of right panel in the figure. Users may test this functionality at http://expressionbrowser.com/arab/displayFeature.jsp?id=1001343.

The T/C View of "16 hr Pseudomonas infection". When PR1, PR2, PR3, PR4 and PR5 were searched and selected on this T/C View, the data points on the MA plot on left panel are labeled with a colored box. The color can be changed by right clicking on the color icon on the lower box of the right panel in this figure. You may test this function at http://expressionbrowser.com/arab/displayPair.jsp?id=2056966.
Gene View and co-responsive genes
The GEB backend data model is a matrix with two dimensions, genes and T/Cs. Users visualize the expression profiles as a slice along either of these two dimensions: the Gene View displays data points of all T/Cs for a selected gene, whereas the T/C View displays data points of all genes for a selected T/C.
Figure 4 illustrates the Gene View. Data points from all T/Cs for a gene are displayed in the MA plot [16, 17]. Here, M, the y-axis, is the log2 ratio of treatment over control (LOG2R) [log2 (treatment intensity) - log2 (control intensity)] and A, the x-axis, is the average log2 intensity of the treatment and control [(log2 (treatment intensity) + log2 (control intensity))/2]. The MA plot provides a quick overview of data points for all T/Cs affecting the selected gene. The data points located in the upper area of the MA plot are 'up-regulation' T/Cs, and those located at lower area are 'down-regulation' T/Cs. Gene View is a cross-experimental display of the expressions of a gene under all experimental conditions currently available in GEB. With the MA plot, users can get a clear overall view of a gene-expression profile without scrolling down the display page, no matter how many data points might be on the plot.
From a GEB MA plot, users can easily view both the LOG2R changes and also the statistical significance of the LOG2R. Each data point is color coded on the basis of the t-test P value that indicates the significance level of its LOG2R. The data points are coded in blue color when P values are lower than 0.01, in green color when P values are between 0.01 and 0.05, and in yellow color when P values are higher than 0.05. The color-coded data points help users know visually significance levels and reliability of the data. For example, if the data point has both a high-fold change (at the top or bottom of the display) and high P values (P > 0.05, yellow color), it suggests that there may be large systematic or experimental errors among replications so that the results should be interpreted cautiously before conclusion are drawn based on such a data point. Therefore, the location and color of the data points on the GEB MA plot give users a clear view of gene expression in both ratio scale and significance level (reliability).
The MA plot is a JPEG image generated by the offline data-processing pipeline. The image is about 60 K in size, with 480 × 480 pixel dimensions, which allows the image to be loaded from host server to users' browser very quickly so that users can rapidly obtain an overall view of the expression profile of a gene. Most importantly, GEB is equipped with highlighting and search functions that allow users to highlight data points by dragging-and-dropping the mouse and to search data by entering keywords. Figure 4 illustrates how to use the "highlighting window" to locate the up-regulation T/Cs on the MA plot. First, users move the "highlighting window" to cover the data points on the upper panel of the MA plot. The users can resize the window, if needed. The two text boxes to the right of the MA plot are used for listing detailed information about the highlighted data points. Users can click the 'Select' button for any T/C on the upper text box and then the selected T/C will be moved to lower text box. At the same time, the selected T/C is also marked on the MA plot with a small rectangle. This two-layer display solution achieves both a quick overview of an expression profile and a detailed view of the selected data points.
Arabidopsis PR-1 gene, a pathogenesis-related gene [19], was used as an example of Gene View in Figure 4. The up-regulation T/Cs selected in Figure 4 are listed in Table 1. A total of 95 T/Cs were selected when 2-fold and P < 0.05 were used as a double cutoff. Among the 95 T/Cs, 44 T/Cs are pathogen treatments, 13 T/Cs are plant defense elicitor treatments, and 14 T/Cs are plant defense-related mutants. These results clearly suggested that the expression of PR-1 was promoted by infections, plant-defense elicitors, and plant defense-related mutations. In previous studies, PR-1 was defined as a pathogenesis-related gene that was coordinately activated by pathogen infection and functioned as an indicator of the defense reaction [20, 21]. The silencing of this gene leads to an increase in extracellular β-(1→3)-glucanase activity at the onset of tobacco defense reactions [22–24]. A decrease in β-(1→3)-glucan deposition in PR-1-silenced lines [22] might cause less deposition of callose that is linked with β-(1→3)-glucan and while the callose deposition is one of the characteristics of defense reactions associated with hypersensitive response of a plant [25]. Morris et al. [26] indicated that chemical induction of maize PR-1 genes increased resistance to downy mildew. The results for PR-1 functions revealed by GEB were impressively consistent with the previous findings. These results strongly suggested that Gene View of GEB would be very useful in gene-function discovery, biomarker validation, and bioprocess identification.
Figure 6 represents a screenshot of "Co-responsive Genes" tab in the PR-1 Gene View (http://www.expressionbrowser.com/arab/displayFeature.jsp?id=1001343&tab=4). The co-responsive relationship of two genes is determined by the following procedure: (1) The up- and down-regulation T/Cs of the two genes are selected using a double cutoff of P < 0.05 and of 2-fold; (2) the overlap T/Cs that have the two genes selected are then used to compute the overlap percentage; (3) the Pearson correlation coefficient is calculated using the LOG2R of overlapped T/Cs; and (4) a relationship index is calculated using the overlap percentage multiplied by the square of the correlation coefficient. The relationship between the two co-responsive genes is computed with ratio data from T/C with only a single factor differing between treatment and control. Therefore, the relationship between co-responsive genes solely reflects the effect of a biological treatment because the variations caused by most other factors are removed. On the other hand, if the relationship between co-expressed genes is computed with intensity data where multiple factors vary (such as tissue and cell type of sample, biological treatment, sampling methods, such as time and location, experimental methods, such as sample storage, mRNA extraction, or microarray dying, and systematic errors), then the relationship between co-expression genes reflects the mixed effects from biological treatment and these multiple factors. In the list of PR-1 co-responsive genes (Figure 6), impressively, many well-known plant defense-related genes, such as EXLB3, PR-2, Chitinase, PR-5 and AGP5, were found. Among them, PR-2 and PR-5 are considered to have a similar function as PR-1 in systemically acquired resistance (SAR) responses [27]. According to a review on the integrated application of online data mining tools by Meier and Gehring [28], PR-1, PR-2 and PR-5 were induced by necrotrophic Botrytis cinerea pathogen. The results shown by GEB are consistent with those from previous studies. The consensus results from multiple experiments in GEB provide reliable clues for gene-expression discoveries.

The co-responsive genes related to PR-1. The co-responsive genes were listed in the order of their relation index to PR-1 genes. The relation index is a product of the overlap percentage (the percentage of overlapped co-regulation T/Cs between PR-1 and the selected gene) and the correlation coefficient (the Pearson correlation coefficient among the overlapped T/Cs). More co-regulation T/Cs can be found at http://expressionbrowser.com/arab/displayFeature.jsp?id=1001343&tab=4.
T/C View and co-regulation T/Cs
Figure 5 represents an example of T/C View of "16 hr Pseudomonas infection." Each data point on the T/C View is the LOG2R of a gene. The MA plot, color codes, two-layer display design, and searching/highlighting functions on the T/C View are exactly the same as those in Gene View described above. The following example shows how to use search function to locate genes in the T/C View. When a string of "PR1 PR2 PR3 PR4 PR5" was used as a search keyword, all genes with any matching word in its annotation are shown in the upper right box (Figure 5). By clicking the 'Select' button on each gene, the gene is moved to the lower box. At the same time, the selected gene is marked on an MA plot with a small rectangle. T/C view provides a condition-centric view of microarray data.
Though different T/Cs may stimulate different sets of genes, any two different T/Cs may co-regulate a set of genes such that they have similar gene-expression signatures. The co-regulation relationship between two T/Cs can be constructed from the similarity of gene-expression signatures of the two T/Cs. If we click the "Co-regulation T/Cs" tab in the T/C View of "16 hr Pseudomonas infection" (http://expressionbrowser.com/arab/displayPair.jsp?id=2056966&tab=4), a total of 199 co-regulation T/Cs are listed in a table ordered by their "relation index" to the "16 hr Pseudomonas infection" T/C. The calculation of relation index between the two T/Cs is described in the footnote in Table 2. The T/C of "24 hr Pseudomonas infection" has the closest relationship (with relation index of 0.623816) to "16 hr Pseudomonas infection." This result is easily understood because they are the same treatment with an 8-hour treatment-time difference. The top 80 (of the 199) co-regulation T/Cs of "16 hr Pseudomonas infection" are listed in Table 2: 29 belong to pathogen-infection, 16 are plant-defense elicitors, and 6 are plant defense-related mutants. It is interesting to note that 3 T/Cs are negatively correlated with the T/C of "16 hr Pseudomonas infection" (Table 2). Two of the three T/Cs are mutants of Enhanced Disease Susceptibility 16 (EDS16) under infection conditions. EDS genes have special function in basal disease resistance to pathogens as well as R genes [29, 30]. Arabidopsis EDS mutants, such as eds1[31] and eds5[32], have lower PR gene-expression level and exhibit higher susceptibility to pathogen infection. The reverse relationship of gene-expression signatures between EDS16 under infection and "16 hr Pseudomonas infection" implies that some pathogen-related genes are either not activated or reduced in EDS16 mutants when they are infected by pathogens. Another T/C negatively correlated with the "16 hr Pseudomonas infection" is caused by "high nitrogen effect". Hoffland et al. [33] reported that high nitrogen application caused higher N concentration in plant tissue, and the effect of tissue N concentration on disease susceptibility was highly pathogen-dependent. They found that disease susceptibility to P. syringae and Oidium lycopersicum was significantly increased with increasing N concentration in tomato tissue [34]. The results obtained from GEB are consistent with the previous independent studies, further suggesting that the results generated by GEB are reliable and the logic/principles implemented in GEB are scientifically sound.
Gene network building has been a hot research topic during the past few years [10, 12, 34, 35]. GEB is not only able to construct gene networks based on the co-responsive relationship described above (Figure 6) but is also able to construct T/C networks based on the co-regulation relationship (Table 2). Another paper will address the details about constructing gene networks and T/C networks in Arabidopsis.
Slide View
The slide view of genes or T/Cs is designed to help users discover changes in multiple genes under various T/Cs or vice versa. Users can make a slide show to compare a set of T/Cs with multiple selected genes. For example, the user can search T/C conditions in Gene View (Figure 4) by typing "cold" in the search box and then selecting three T/C conditions with cold treatment of 12 hr, 6 hr, and 3 hr from the upper right box to the lower right box. After selecting the three "cold" conditions, the user can also search another three non-related T/Cs, such as "drought," "UV-B" and "wounding" with the same procedure. After the six necessary T/C conditions are selected, the user can click the "[slide]" link and then the six MA plots of T/Cs are shown as slides (Figure 7). In Figure 7A, the user highlights a certain number of genes by dragging, dropping and resizing the "highlighting window." A total of 51 genes with at least 30-fold increase (LOG2R > 4.9) in 12-hr cold condition are selected. To see how the selected genes are changed in other T/Cs, click the "next slide" arrow, and the next slide will appear. The selected genes in the first slide are still highlighted but the positions of the selected genes are changed in different slides. With this slide show, users are able to see the change of these 51 selected genes in different T/Cs. Figures 7B to 7F reveal changes in the selected genes in the T/Cs with treatments of 6-hr cold, 3-hr cold, 12-hr drought, 12-hr UV-B, and 12-hr wounding, respectively. These slides clearly demonstrate that the selected genes had highest fold changes under 12-hour cold treatment (Figure 7A). The fold-changes decreased in 6-hr (Figure 7B) and 3-hr (Figure 7C) cold treatments. The positions of the 51 selected genes in treatments of drought (Figure 7D), UV-B (Figure 7E) and wounding (Figure 7F) showed less similarity to "12 hr cold treatment". The Slide View is a very simple and powerful visualization tool for scientists to compare their candidate genes and see how the genes behave differently in the T/Cs across studies.

A sample of Slide View for six T/Cs. (3 cold treatments, 1 drought treatment, 1 UV-B treatment, and 1 wounding treatment). Users may test this function at http://www.expressionbrowser.com/arab/displayPairSlides.jsp?id=2055336&id=2055335&id=2055334&id=2055599&id=2055979&id=2056077.
Experiment View
Experiment View shows experiment title, description/design, lab information, samples, sample sets, biological replicates, and the definitions of T/Cs. This view helps users understand the data in detail. For example, the contents of the Experiment View of "Pathogen Series: Pseudomonas half leaf injection" can be seen at the following link http://expressionbrowser.com/arab/displayExperiment.jsp?id=2020113. There are three tabs in the Experiment View. The first tab, called "Details," displays experiment title, description, and other detailed information of the experiment. The second tab "T/C" contains information about the T/Cs in the experiment. The third tab "Samples and Data" contains information about all sample sets, samples and raw data files. Users can download raw microarray data files through the "Samples and Data" tab and then input these raw data into other microarray data-analysis software to analyze the data and to validate the results obtained from GEB.
Conclusions
GEB is composed of a data extraction tool, a microarray data-management system, a data annotation tool, a data-processing pipeline, and a search & visualization tool. The heterogeneity of diverse experimental designs has been greatly mitigated by re-organizing different experimental treatment levels into T/Cs so that cross-experimental data integration is easily achieved. GEB separates data processing from interactive display. It pre-processes data and generates data plot images, and then displays the processed data with a web2.0-based interactive user-interface, according to users' requests. This design allows heavy computing to be done offline, and thus allows a large number of data points to be queried quickly and displayed interactively in real-time. GEB displays all data points in one view so that users do not need to scroll down display pages to obtain the trend or pattern of gene expressions from all data points. The highlighting and searching functions in Gene View, T/C View, and Slide View greatly facilitate dynamically exploring the data points based on users' interests. As an additional strategy to improve usability, all raw data and calculated data in GEB are accessible via a full-text search engine. GEB also computes relations of co-regulation T/Cs and co-responsive genes. These relations are the foundation for building gene networks and T/C networks.
Availability and requirements
-
Project Name: Gene Expression Browser (GEB)
-
Public web service: http://www.ExpressionBrowser.com Free and no registration.
-
Programming Language: Java, R
-
Database: MySQL
-
Software License: The software license is owned by GeneExp. GeneExp grants free licenses to non-profit organizations and general licenses to commercial organizations.
-
License request: [email protected]
Abbreviations
- A:
-
Average Log2 Intensity
- AJAX:
-
Asynchronous JavaScript and XML
- ATH1:
-
A light-regulated Arabidopsis thaliana homeobox 1 gene
- BTH:
-
Benzothiadiazole
- CC:
-
Correlation Coefficient
- CIBEX:
-
Center for Information Biology Gene Expression Database
- CPU:
-
Central Processing Unit
- EDS:
-
Enhanced Disease Susceptibility
- EBI:
-
The European Bioinformatics Institute
- GDS:
-
Granite Data Services
- GEB:
-
Gene Express Browser
- GEO:
-
Gene Expression Omnibus
- JPEG:
-
Joint Photographic Experts Group
- LOG2R:
-
Log2 Ratio of Treatment over Control
- MA plot:
-
a Quick Overview of Intensity-dependent Ratio of Microarray Data
- M:
-
LOG2R
- N:
-
Nitrogen
- NASC:
-
Nottingham Arabidopsis Stock Center
- NCBI:
-
The National Center for Biotechnology Information
- NIG:
-
The National Institute of Genetics
- PR gene:
-
Pathogenesis-related gene
- R gene:
-
Resistance genes
- RI:
-
Relation Index
- RMA:
-
Robust Multichip Average
- SA:
-
Salicylic Acid
- SAR:
-
Systemic Acquired Resistance
- TAIR:
-
Texas Association for Institutional Research
- T/C:
-
Treatment over Control
- %CV:
-
Percentage Coefficient of Variation
- OP:
-
Overlapping Percentage
- UV-B:
-
Ultraviolet-B Radiation.
References
Chen R, Mallelwar R, Thosar A, Venkatasubrahmanyam S, Butte AJ: GeneChaser: identifying all biological and clinical conditions in which genes of interest are differentially expressed. BMC Bioinformatics 2008, 9: 548. 10.1186/1471-2105-9-548
Penkett CJ, Bähler J: Navigating public microarray databases. Comparative and Functional Genomics 2004, 5(6–7):471–479. 10.1002/cfg.427
Edgar R, Domrachev M, Lash AE: Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research 2002, 30(1):207–210. 10.1093/nar/30.1.207
Brazma A, Sarkans U: Gene Expression Databases. In Nature encyclopedia of the human genome. Edited by: Cooper D. Nature Publishing Group, London; 2003:628–632.
Rocca-Serra P, Brazma A, Parkinson H, Sarkans U, Shojatalab M, Contrino S, Vilo J, Abeygunawardena N, Mukherjee G, Holloway E, Kapushesky M, Kemmeren P, Lara GG, Oezcimen A, Sansone S: ArrayExpress: a public database of gene expression data at EBI. Comptes Rendus Biologies 2003, 326(10):1075–1078. 10.1016/j.crvi.2003.09.026
Ikeo K, Ishi-i J, Tamura T, Gojobori T, Tateno Y: CIBEX: center for information biology gene expression database. Comptes Rendus Biologies 2003, 326(10):1079–1082. 10.1016/j.crvi.2003.09.034
Trivedi P, Edawards JW, Wang JL, Gadbury GL, Srinivasasainagenda V, Zakharkin SO, Kim K, Mehta T, Brand JPL, Patki A, Page GP, Allison DB: HDBStat! A platform-independent software suite for statistical analysis of high dimensional biology data. BMC Bioinformatics 2005, 6: 86. 10.1186/1471-2105-6-86
Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W: GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiology 2004, 136(1):2621–2632. 10.1104/pp.104.046367
Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S: NASCArrays: a repository for microarray data generated by NASC's transcriptomics service. Nucleic Acids Research 2004, (32 Database):D575-D577. 10.1093/nar/gkh133
Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Scholkopf B, Weigel D, Lohmann JU: A gene expression map of Arabidopsis thaliana development. Nature Genetics 2005, 37(5):501–506. 10.1038/ng1543
Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D'Angelo C, Bornberg-Bauer E, Kudla J, Harter K: The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. The Plant Journal 2007, 50(2):347–363. 10.1111/j.1365-313X.2007.03052.x
Ma S, Gong Q, Bohnert HJ: An Arabidopsis gene network based on the graphical Gaussian model. Genome Research 2007, 17(11):1614–1625. 10.1101/gr.6911207
Koornneef M, Scheres B: Arabidopsis thaliana as an experimental organism. Encyclopedia of Life Sciences 2001, 1–6.
Agrawal GK, Yonekura M, Iwahashi Y, Iwahashi H, Rakwal R: System, trends and perspectives of proteomics in dicot plants. Part I: Technologies in proteome establishment. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences 2005, 815(1–2):109–123. 10.1016/j.jchromb.2004.11.024
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4(2):249–264. 10.1093/biostatistics/4.2.249
Dudoit S, Yang YH, Callow MJ, Speed TP: Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statistica Sinica 2002, 12(1):111–140.
Bolstad BM, Irizarry RA, Astrand M, Speed TP: A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19(2):185–193. 10.1093/bioinformatics/19.2.185
Garrett JJ: Ajax: A new approach to web application.2005. [http://www.adaptivepath.com/ideas/essays/archives/000385.php]
Cao H, Glazebrook J, Clark JD, Volko S, Dong X: The Arabidopsis NPR1 gene that controls systemic acquired resistance encodes a novel protein containing ankyrin repeats. Cell 1997, 88(1):57–63. 10.1016/S0092-8674(00)81858-9
Durner J, Shah J, Klessig DF: Salicylic acid and disease resistance in plants. Trends in Plant Science 1997, 2(7):266–274. 10.1016/S1360-1385(97)86349-2
De Vos M, Van Zaanen W, Koornneef A, Korzelius JP, Dicke M, Van Loon LC, Pieterse CM: Herbivore-induced resistance against microbial pathogens in Arabidopsis . Plant Physiology 2006, 142(1):352–363. 10.1104/pp.106.083907
Rivière MP, Marais A, Ponchet M, Willats W, Galiana E: Silencing of acidic pathogenesis-related PR-1 genes increases extracellular beta-(1→3)-glucanase activity at the onset of tobacco defence reactions. Journal of Experimental Botany 2008, 59(6):1225–1239. 10.1093/jxb/ern044
Bowles JD: Defense-related proteins in higher plants. Annual Review of Biochemistry 1990, 59: 873–907. 10.1146/annurev.bi.59.070190.004301
Boller T: Antimicrobial functions of the plant hydrolases chitinase and β-1,3-glucanase. In Developments In Plant Pathology. Edited by: Fritig B, Legrand M. Springer, New York; 1993:391–400.
Stone BA, Clarke AE: (1,3)-β-glucans in plant host-pathogen interactions. In Chemistry and biology of (1,3)-β-glucan. La Trobe University Press, Bundoora, Australia; 1992:491–512.
Morris SW, Vernooij B, Titatarn S, Starrett M, Thomas S, Wiltse CC, Frederiksen RA, Bhandhufalck A, Hulbert S, Uknes S: Induced resistance responses in maize. Molecular Plant Microbe Interactions 1998, 11(7):643–658. 10.1094/MPMI.1998.11.7.643
Wildermuth MC, Dewdney J, Wu G, Ausubel FM: Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 2001, 414(6863):562–565. 10.1038/35107108
Meier S, Gehring C: A guide to the integrated application of on-line data mining tools for the inference of gene functions at the systems level. Biotechnology Journal 2008, 3(11):1375–1387. 10.1002/biot.200800142
Glazebrook J: Genes controlling expression of defense responses in Arabidopsis --2001 status. Current Opinion in Plant Biology 2001, 4(4):301–308. 10.1016/S1369-5266(00)00177-1
Venugopal1 SC, Jeong R, Mandal MK, Zhu SF, Chandra-Shekara1 AC, Xia Y, Hersh M, Stromberg AJ, Navarre D, Kachroo A, Kachroo P: Enhanced Disease Susceptibility 1 and Salicylic Acid Act Redundantly to Regulate Resistance Gene-Mediated Signaling. PLoS Genet 2009, 5(7):e1000545. 10.1371/journal.pgen.1000545
Falk A, Feys BJ, Frost LN, Jones JD, Daniels MJ, Parker JE: EDS1, an essential component of R gene-mediated disease resistance in Arabidopsis has homology to eukaryotic lipases. Proceedings of the National Academy of Sciences of the United States of America 1999, 96(6):3292–3297. 10.1073/pnas.96.6.3292
Nawrath C, Heck S, Parinthawong N, Metraux JP: EDS5, an essential component of salicylic acid-dependent signaling for disease resistance in Arabidopsis , is a member of the MATE transport family. Plant Cell 2002, 14(1):275–286. 10.1105/tpc.010376
Hoffland E, Jeger MJ, Van Beusichem ML: Effect of nitrogen supply rate on disease resistnce in tomato depends on the pathogen. Plant and Soil 2000, 218(1–2):239–248. 10.1023/A:1014960507981
Freeman TC, Goldovsky L, Brosch M, Dongen SV, Maziere P, Grocock RJ, Freilich S, Thornton J, Enright AJ: Construction, visualization, and clustering of transcription networks from microarray expression data. PLoS Computational Biology 2007, 3(10):e206. 10.1371/journal.pcbi.0030206
Theocharidis T, Dongen SV, Enright AJ, Freeman T: Network visualization and analysis of gene expression data using BioLayout Express3D. Nature Protocols 2009, 4(10):1535–1550. 10.1038/nprot.2009.177
Acknowledgements
We thank Dr Michael Gibson for creative discussions on the design of software architecture and user interfaces and for critically reading this paper. We also thank Professor Zhiyong Wang for coining the term "co-responsive genes." Arabidopsis data input and annotation are supported by the Yunnan Advanced Talent Introduction Project Foundation (20080A006), China.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MZ, XF, ST proposed software requirements. MZ, YZ, XF, MSK did software specification and design. YZ developed statistical protocols. MZ designed database schema and developed computational algorithms and the software. LL, LY, ST, JT, WY, YA tested the software application and wrote the manual. MZ downloaded and processed raw microarray data from GEO and NASC. LL, LY, ST, JT, WY annotated the data and nominated the T/Cs. MZ, YZ, XF, ST, MSK, YA drafted the manuscript. MZ, YZ, XF, LL, LY, ST, JT, WY, MSK wrote different parts of the manuscript. MZ, YZ, XF, MSK, YA assembled all parts written by different authors together into this manuscript. All authors read and approved this manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, M., Zhang, Y., Liu, L. et al. Gene Expression Browser: large-scale and cross-experiment microarray data integration, management, search & visualization. BMC Bioinformatics 11, 433 (2010). https://doi.org/10.1186/1471-2105-11-433
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-11-433