Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies.
![]() Free full text in Europe PMC
Free full text in Europe PMC
Abstract
Free full text

Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies
Abstract
Next-generation sequencing technologies can be used to analyse genetically heterogeneous samples at unprecedented detail. The high coverage achievable with these methods enables the detection of many low-frequency variants. However, sequencing errors complicate the analysis of mixed populations and result in inflated estimates of genetic diversity. We developed a probabilistic Bayesian approach to minimize the effect of errors on the detection of minority variants. We applied it to pyrosequencing data obtained from a 1.5-kb-fragment of the HIV-1 gag/pol gene in two control and two clinical samples. The effect of PCR amplification was analysed. Error correction resulted in a two- and five-fold decrease of the pyrosequencing base substitution rate, from 0.05% to 0.03% and from 0.25% to 0.05% in the non-PCR and PCR-amplified samples, respectively. We were able to detect viral clones as rare as 0.1% with perfect sequence reconstruction. Probabilistic haplotype inference outperforms the counting-based calling method in both precision and recall. Genetic diversity observed within and between two clinical samples resulted in various patterns of phenotypic drug resistance and suggests a close epidemiological link. We conclude that pyrosequencing can be used to investigate genetically diverse samples with high accuracy if technical errors are properly treated.
INTRODUCTION
Recent technological advances have drastically decreased the time and the cost required to obtain DNA sequences (1). Several next-generation sequencing (NGS), or deep sequencing, platforms are available now that can read millions of base pairs in a more cost-effective and faster way than traditional Sanger sequencing (2). NGS is applied in de novo genome sequencing projects (3), as well as in targeted resequencing studies, for example, of tumours (4), in epigenetic studies (5) and in transcriptome analysis (6). In this article, we demonstrate how NGS can be used to detect low-frequency variants in genetically heterogeneous samples such as those obtained from HIV-infected patients.
In general, the population structure of infectious pathogens is highly relevant because genetic pathogen diversity is often associated with disease progression, poor prognosis and treatment failure. RNA viruses, such as HIV or influenza, are prominent examples. The low fidelity of their viral polymerases, which lack classic proofreading mechanisms, is responsible for the continuous production of mutated viral copies. Many mutations are maintained on clones in proportion related to their fitness. This mutant spectrum, referred to as a viral quasispecies (7), allows the virus to rapidly adapt to fluctuating environments (8,9).
Without additional experimental effort, such as individual cloning, Sanger capillary sequencing can only determine the consensus sequence of a mixed sample, and mutations can only be detected if their frequency exceeds a threshold of ~20%. If two or more loci display variation, information on whether and how often these variants occur on the same DNA molecule is lost. NGS can overcome these limitations by directly sequencing the mixed sample at high coverage. Every read obtained in this manner represents a contiguous fragment of DNA from a single molecule in the DNA library of the sample. Therefore, the set of reads provides a statistical sample of the DNA library and it can be used to make inference about the genetic structure of the population.
The potential for NGS to detect low-frequency variants has been noted early on in the context of viral infections. Pyrosequencing detected low-frequency mutations conferring resistance to antiviral drugs in hepatitis B virus (10,11) and in HIV (12–16), and found mixed infections with different influenza strains (17,18).
Despite these early successful case studies, application of NGS to resolve the population structure of pathogens remains challenging, chiefly because of the sequencing error rate. A consequence of the high error rate is the risk of considering a technical error as a low-frequency variant. For example, with a uniform error rate of 0.25% per base pair and an average read length of 350 bp, ~58% of the reads obtained in an experiment are expected to be contaminated with at least one sequencing error. Thus, the genetic diversity of the sample would be vastly overestimated based on the raw data alone. To avoid these huge numbers of false positives, some ad hoc strategies have been proposed to discard reads that are much shorter than average, contain ambiguous characters, or induce frameshifts in the translation (19,20).
bp, ~58% of the reads obtained in an experiment are expected to be contaminated with at least one sequencing error. Thus, the genetic diversity of the sample would be vastly overestimated based on the raw data alone. To avoid these huge numbers of false positives, some ad hoc strategies have been proposed to discard reads that are much shorter than average, contain ambiguous characters, or induce frameshifts in the translation (19,20).
Statistically, measurement noise can be distinguished from real variation in a deep sequencing experiment, if variants that are sequenced more than once can be identified as groups (clusters) of reads that are more similar to each other than to reads in other groups (Supplementary Figure S1). Here, we use a Bayesian statistical modelling approach to read error correction that can be regarded as a probabilistic clustering method (21). The main advantages of our approach are: (i) that an independent estimate of the error rate is not necessary; (ii) that the number of haplotypes does not need to be specified in advance; and (iii) that we generate a full probabilistic clustering solution which contains all information about the uncertainty associated with it, including the estimated error rate, the estimated number of haplotypes, their DNA sequence composition and the assignment of reads to haplotypes. This algorithm, together with other tools, is implemented in the open source software package ShoRAH (www.cbg.ethz.ch/software/shorah).
We present a comprehensive analysis of this method and assess its performance in error correction, haplotype reconstruction and haplotype frequency estimation. We applied the ShoRAH algorithm to read data from four HIV samples obtained with the 454/Roche GS FLX Titanium platform (Table 1). Two samples define control experiments and consist of 10 different clonal isolates of the HIV pol gene, mixed in different proportions. To assess the effect of PCR amplification, another confounding factor, one of the control samples was PCR-amplified from an aliquot of the other before pyrosequencing. The remaining two samples were derived from HIV patients suspected to be part of the same infection chain.
Table 1.
Next-generation sequencing experiments
| Experiment | Sample | Coverage | Indels (%) | Mismatches (%) | 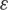 (%) (%) | nt hap |
|---|---|---|---|---|---|---|
| Control | Non-PCR | 2110 | 1.0 | 0.05 | 0.35–0.60 | 10 |
| Control | PCR | 6030 | 1.0 | 0.25 | 0.40–0.45 | 10 |
| Clinical | Patient 1 | 2100 | 0.44 | – | 1.1 | 13 |
| Clinical | Patient 2 | 6240 | 0.42 | – | 0.8 | 15 |
Type of experiment, coverage, error rates, estimates for the parameter 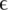 and number of haplotypes (nt hap) are reported for all experiments.
and number of haplotypes (nt hap) are reported for all experiments. 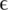 represents the probability that a position in one of the reads in the multiple sequence alignment is wrong. The number of haplotypes in the clinical samples is the number of distinct haplotypes that were detected.
represents the probability that a position in one of the reads in the multiple sequence alignment is wrong. The number of haplotypes in the clinical samples is the number of distinct haplotypes that were detected.
The control experiments allow for a hard assessment of the performance of the computational haplotype inference method, because the reads can be mapped directly to the original clones, which are the only true haplotypes in the mixtures. We demonstrate that haplotypes at frequencies as low as 0.1% can be detected reliably and that their estimated frequencies show high agreement with the expected frequencies. Haplotype reconstruction based on probabilistic clustering is shown to outperform, in both precision and recall, ad hoc methods based on a minimal number of required observations.
In the clinical samples obtained from two infected patients, we found several low-frequency mutations, which were invisible to Sanger sequencing. The pattern of observed mutations was used to analyse the level of resistance of individual variants to several protease inhibitors (PIs). Both viral populations display intra-host genetic heterogeneity that resulted in predicted phenotypic diversity of drug resistance. In particular, we found minority drug resistant variants that, as it has been repeatedly observed, can affect treatment outcome (11,14–16,22).
It seems obvious that high-coverage NGS can detect low-frequency variants in the pathogen populations. In practice, however, the feasibility of this approach depends critically on correcting sequencing errors. Probabilistic read clustering as implemented in ShoRAH provides a robust error correction method and it can be used to estimate the structure of pathogen populations in short genomic regions.
MATERIALS AND METHODS
Experiments
Sample preparation
For the generation of a defined clone mixture, amplicons of the partial gag/pol-gene were produced in the context of routine genotypic HIV drug resistance testing. Briefly, viral RNA was isolated from plasma of HIV infected patients using QIAamp viral RNA mini kit (Qiagen, Hilden, Germany) according to the manufacturers protocol. Reverse transcription and polymerase chain reaction were carried out using OneStep RT–PCR kit (Qiagen, Hilden, Germany) and primers
1RES, 5′-GAAGAAATGATGACAGCATGTCAGGG-3′ (nt 1819–1844 numbered according to HXB2 reference genome) and
2RES, 5′-TAATTTATCTACTTGTTCATTTCCTCCAAT-3′ (nt 4173–4202).
Nested PCR was carried out with HotStarTaq (Qiagen, Hilden, Germany) and the following inner primer pair:
RES3, 5′-AGACAGGCTAATTTTTTAGGGA-3′ (nt 2074–2095) and
RES4, 5′-ATGGYTCTTGATAAATTTGATATGTCC-3′ (nt 3559–3585).
The 1.5-kb PCR product was purified by using the QIAquick spin PCR purification kit (Qiagen, Hilden, Germany). Standard Sanger sequencing of the HIV-1 pol region was done by using ABI Prism 3730 capillary sequencer (Applied Biosystems, Foster City, CA, USA). PCR-products from 10 different subtype-B clinical isolates were cloned into pCRII-TOPO (Invitrogen, Carlsbad, CA, USA). After control sequencing and propagation, the inserts were excised from the vector and restriction enzymes SpeI and NotI. To analyse the impact of the PCR on the error rate an aliquot containing 100000 copies of the fragment was used as the template in a single round PCR reaction using primers RES3 and RES4 (PCR-amplified sample). Samples from Patients 1 and 2 were generated by PCR from clinical specimen as described above. All PCR product were purified using QIAamp PCR purification kit (Qiagen, Hilden, Germany) until further processing.
Massively parallel sequencing
For the library preparation all four samples were nebulized according to 454 shotgun protocol (Roche/454-Life sciences, Branford, CT, USA). Fragmented DNA was purified using AMPure SPRI beads (Agencourt) to remove fragments <400bp. The purified fragmented DNA was further processed according to the 454 FLX Titanium Library construction kit and protocol (Roche/454-Life Sciences, Branford, CT, USA) to ligate multiple identifier (MID) adaptors specific to the Titanium sequencing chemistry. The resulting single-stranded DNA library was assessed for size distribution using the RNA 6000 Pico chip on the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and quantified using the Ribogreen RNA Quantitation Kit (Invitrogen) on a Fluorometer (FLUO-star Omega, BMG Labtech Offenburg, Germany). Emulsion PCR (emPCR) and titration by enrichment at 0.5, 1, 2 and 4 copies per bead (cpb) was carried out according to the 454 Titanium emPCR protocol to determine the optimal ratio of the library DNA fragments to emPCR beads. Large volume emulsions were set up at the optimal ratio of 2 DNA cpb. The sequencing run was carried out on a 2/4 picotiter plate using the Roche/454 Genome Sequencer FLX with the 454 Titanium update.
Data analysis
Sample selection
In the first experiment, we mixed 10 different 1.5-kbp HIV-1 clonal fragments of the gag/pol gene at different proportions between 0.3% and 30%. In the analysis, we focused on a 1245-bp long region including the complete viral protease and part of the reverse transcriptase from position 2253 to 3497. The clones were chosen such that their mutual distance be in a narrow range (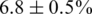 ). Nevertheless, some regions are more conserved than others, so when the haplotypes are compared on localized windows, the diversity varies between 2% and 12%, see Supplementary Figures 4 and 5. The original and the PCR-amplified mixtures were ligated with MID barcodes and sequenced. Among the 63019 reads obtained, the MIDs assigned 16540 reads to Sample 1 (non-PCR-amplified) and 45973 to Sample 2 (PCR-amplified). We decided not to assign the remaining reads because they presented two or more mismatches in the barcode, and we regarded this as an indication of a low-quality read, although we cannot exclude that the mismatches were due to mistakes in the production of MIDs. In the second experiment, plasma samples from two HIV infected patients were analysed. The same genomic region as in the control experiment was isolated, ligated to barcodes and sequenced. In this experiment, 48285 reads were obtained, 12626 assigned to Patient 1 and 35088 to Patient 2.
). Nevertheless, some regions are more conserved than others, so when the haplotypes are compared on localized windows, the diversity varies between 2% and 12%, see Supplementary Figures 4 and 5. The original and the PCR-amplified mixtures were ligated with MID barcodes and sequenced. Among the 63019 reads obtained, the MIDs assigned 16540 reads to Sample 1 (non-PCR-amplified) and 45973 to Sample 2 (PCR-amplified). We decided not to assign the remaining reads because they presented two or more mismatches in the barcode, and we regarded this as an indication of a low-quality read, although we cannot exclude that the mismatches were due to mistakes in the production of MIDs. In the second experiment, plasma samples from two HIV infected patients were analysed. The same genomic region as in the control experiment was isolated, ligated to barcodes and sequenced. In this experiment, 48285 reads were obtained, 12626 assigned to Patient 1 and 35088 to Patient 2.
Preprocessing
For all four samples, we removed low-quality reads defined as having a phred score below 10 at one or more sequence positions. This filtering step selected 10907 and 26814 reads for the non-PCR and PCR control samples, and 7985 and 23284 reads for Patients 1 and 2, respectively. The average read length was 340bp for the control samples and 380bp for the clinical samples (Table 1). After the filtering step, we built a multiple sequence alignment (MSA) of the reads by padding the gaps obtained in pairwise alignments of the reads to the reference genome, because off-the-shelf MSA software is impractical in this setting (23). The MSA was subsequently split into regions of width approximately equal to the read length, and the correction algorithm was applied to each region.
Haplotype reconstruction and error correction
Every time a variation is observed in the reads, the possibility that this is a technical error rather than a true biological variation must be considered. Due to the high coverage of NGS, we exploited the power gained from multiple independent observations. We used clustering to group reads by their similarity and interpreted an optimal clustering as follows. Each cluster consists of exactly those reads that originate from the same haplotype. The cluster centre corresponds to the haplotype sequence and the cluster size is proportional to its frequency. With this interpretation, sequencing errors can be corrected by removing any variation within read clusters and haplotypes can be reconstructed as cluster centres.
The most difficult task in clustering is usually to find the number of clusters that best explain the data (24). In our context, given a set of reads, this number depends on the error rate, that should be measured in a different experiment (25). To circumvent this difficulty, we developed a probabilistic generative model for the sequencing of error-prone reads from a mixed sample. In our approach, the number of haplotypes and the technical error rate are not fixed a priori, rather they are parameters to be estimated together with the biological diversity. The statistical model used to explore different numbers of clusters employs a non-parametric prior distribution called Dirichlet process mixture (26). In a Bayesian fashion, we compute the posterior distribution of the assignment of reads to haplotypes, the identity of these haplotypes, and the parameters controlling the error rate and the biological diversity, given the observed reads. Due to the complicated form of this probability distribution, these estimates cannot be derived analytically. We used Gibbs sampling, a Markov chain Monte Carlo (MCMC) algorithm, to sample from the posterior distribution of these quantities.
This model, together with other tools, is implemented in the software ShoRAH (21). A detailed mathematical description of the method and an assessment of its performance on simulated data has been presented elsewhere (26).
Confidence levels
The probabilistic nature of the clustering algorithm allows for estimating the reliability of predictions. The sampling method we devised explores different configurations of the model parameters, including the haplotype sequences. The fraction of iterations a haplotype is reported estimates the posterior probability of the existence of that haplotype. This posterior provides a confidence level for the haplotype, and in general, we report only those haplotypes with confidence value (posterior probability) greater than 0.9.
Control experiments
For the control experiments, we aligned the reads to all original sequences and assigned them to the best matched if there was no ambiguity in the assignment (the difference between the identities with the best match and the second best match had to be 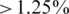 ). We could then estimate the frequencies of the haplotypes, as the fraction of reads assigned to each one, and the sequencing errors from the number of discrepant bases between each read and their closest haplotype. The haplotype frequencies of the two control samples with and without PCR amplification are reported in Supplementary Figure 2. Some deviations are observed, especially for low frequencies, which indicate a selective amplification bias during PCR. Thus, PCR amplification is critical to estimating haplotype frequencies in mixed samples. Haplotype frequencies are also affected by stochastic sampling effects, if the coverage is non-uniform and frequencies are estimated at a local level. Therefore, we locally re-estimated the expected frequencies by re-aligning the read segments in every window of the MSA to the original clone segments of the respective genomic region. These estimates were considered the ground truth and compared to the output of the ShoRAH algorithm.
). We could then estimate the frequencies of the haplotypes, as the fraction of reads assigned to each one, and the sequencing errors from the number of discrepant bases between each read and their closest haplotype. The haplotype frequencies of the two control samples with and without PCR amplification are reported in Supplementary Figure 2. Some deviations are observed, especially for low frequencies, which indicate a selective amplification bias during PCR. Thus, PCR amplification is critical to estimating haplotype frequencies in mixed samples. Haplotype frequencies are also affected by stochastic sampling effects, if the coverage is non-uniform and frequencies are estimated at a local level. Therefore, we locally re-estimated the expected frequencies by re-aligning the read segments in every window of the MSA to the original clone segments of the respective genomic region. These estimates were considered the ground truth and compared to the output of the ShoRAH algorithm.
In the precision–recall analysis, we compared the performance of the cut-off method (in which a minimum number of reads must support a haplotype to call it true) with the probabilistic clustering results. In the cut-off method, the minimum threshold is varied between one and the maximum number of reads in order to draw the precision–recall graph. Similarly, a threshold on the confidence value defines the positive and negative haplotypes reconstructed by the clustering algorithm. The inferred haplotypes were aligned to all sequences, and they were considered correctly inferred if they matched one of the sequences with at most one mismatch. Precision was defined as the ratio of the number of matched sequences (out of the 10 present) over the total number of reconstructed haplotypes. Recall was the ratio of the number of matched sequences over the total number of real sequences (10).
Clinical samples
Phenotypic drug resistance for the reconstructed haplotypes has been predicted with a support vector machine model with linear kernel trained on the data in the Stanford HIV Drug Resistance Database (27) as described in (28) and (29). The coordinates of the points on the plot representing the inferred haplotypes have been determined with a multi-dimensional-scaling technique such that their distance reflects the distance between amino acid sequences.
RESULTS
We used deep sequencing to infer low-frequency variants in a mixed population with high precision, despite the error rate that is typically associated with this technology. To achieve this result, we used ShoRAH, a Bayesian probabilistic clustering method to identify biological variation and to filter out technical errors. The output of the algorithm is a list of haplotypes with a confidence value and an estimate of its frequency (Supplementary Figure S1). In Figure 1, one example of the distribution of these posteriors is shown for a region of the control sample with 10 haplotypes. We observe that about 10 haplotypes have a high posterior probability, while for the 5 additional haplotypes the confidence values quickly drop to negligible levels. For the haplotypes reported we also estimated the frequencies. In particular, low-frequency variants can have high confidence levels, emphasising the benefit of a full probabilistic model over cut-off-based haplotype calling.

Posterior probability of reconstructed haplotypes. The algorithm computes posterior probabilities for the inferred haplotypes and their frequency given the observed reads. The figure shows (for window 3 in the PCR-amplified control experiment) the posterior of the haplotype frequencies in a box-plot (red box-plots) and the posterior of the reconstructed haplotype sequences (blue circles). In most cases, the box-plot height (lower-upper quartile) is invisible on this scale, because the clustering assignment is stable and the number of reads assigned to the cluster in the sampling does not change. The posterior distribution for the frequency is then very peaked. The figure reports estimates for the haplotypes reported by the algorithm without further processing. With additional analysis one finds that haplotypes 12 and 13 differ by one gap only in a homopolymeric region, and that their posterior probabilities sum up to one. Moreover, haplotype 1 consists of four reads and is the result of a recombination event between two of the original haplotypes.
PCR errors
Error rate of pyrosequencing
We estimated the error rate of the NGS procedure from the number of discrepant bases in the alignment of each read with its best match among the original clones (see ‘Materials and Methods’ section). Excluding indels, the base substitution error rate for the non-PCR sample was 0.05%, while for the PCR-amplified sample it was 0.25%. The PCR-associated increase of the error rate was highly significant (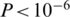 , Wilcoxon rank-sum test).
, Wilcoxon rank-sum test).
Recombination
PCR can not only introduce base substitution errors, but when used to amplify a mixture of heterogeneous templates, it may also produce cross-overs among templates (33). To estimate the amount of this artificial recombination, we analysed the read data from the two control samples with the software Recco (34). For each read, we determined whether it is better explained as resulting from mutations and indels in a single haplotype, or from recombination between two haplotypes and fewer mutations and indels.
The amount of mutations that can be saved by invoking recombination to explain the observed read is a measure of the likelihood of a recombination event. The result of this analysis is reported in Supplementary Figure S3. While for the non-PCR-amplified sample, the number of mutations that could be saved is always lower than five, in the PCR-amplified sample, up to 16 possible savings are observed. In 1.9% of reads from the PCR-amplified sample, we observed more than five savings, indicating PCR-associated cross-overs.
Error correction and haplotype reconstruction
Detection of haplotypes
The high coverage of NGS promises increased sensitivity to detect haplotypes in a mixture, but PCR and sequencing errors can induce large false positive rates without careful analysis of the read data. Using the clonal mixture samples, we compared the ability to detect the 10 real haplotypes between our probabilistic clustering method and the baseline cut-off method. A low cut-off results in the detection of many haplotypes at the cost of many false positives, while a large cut-off avoids false alarms at the cost of missing true variants. For example, requiring only at least two observations of each haplotype (i.e. cut-off 2), we found between 7 and 37 haplotypes in the non-PCR sample and between 80 and 235 haplotypes in the PCR-amplified sample. For a cut-off of 20 observations, between 0 and 7 and between 5 and 16 haplotypes were called for the non-PCR and PCR sample, respectively. Some manufacturers recommend a cut-off of 50 observations (35). For this cut-off, between 0 and 5 haplotypes were found in the non-PCR-amplified sample and between 3 and 10 were found in the PCR-amplified one.
The choice of an optimal cut-off does not only depend on the trade-off between precision and recall, but also on specific parameters such as coverage, error rate and genetic diversity of the sample, some of which are unknown in any real application. The Bayesian clustering approach avoids this problem and instead estimates these parameters directly from the data and reports the confidence level for each reported haplotype.
We assessed haplotype reconstruction performance in a precision–recall analysis over the entire range of possible cut-offs and posterior probabilities. Precision is the fraction of true haplotypes among all called haplotypes and recall is the fraction of called haplotypes among all true haplotypes. Figure 2 shows the aggregate result of this analysis for all windows of the MSA. The cut-off method performs poorly for all choices of the cut-off value and can achieve high recall (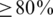 ) only at the expense of very low precision (
) only at the expense of very low precision (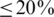 ) and vice versa. For example, a cut-off of 50 observations, results in 80% precision, but only <40% recall (Figure 2, red arrows). The probabilistic clustering method outperforms the cut-off method for any choice of the haplotype confidence level. Overall, the performance is less sensitive to this value and many choices result in haplotype reconstruction with both precision and recall
) and vice versa. For example, a cut-off of 50 observations, results in 80% precision, but only <40% recall (Figure 2, red arrows). The probabilistic clustering method outperforms the cut-off method for any choice of the haplotype confidence level. Overall, the performance is less sensitive to this value and many choices result in haplotype reconstruction with both precision and recall 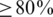 . In general, this method is conservative in the sense that for all confidence thresholds a called haplotype is likely to be a true haplotype, especially for thresholds of 0.9 and higher. For both methods, performance is slightly better on the non-PCR sample than on the PCR sample.
. In general, this method is conservative in the sense that for all confidence thresholds a called haplotype is likely to be a true haplotype, especially for thresholds of 0.9 and higher. For both methods, performance is slightly better on the non-PCR sample than on the PCR sample.

Precision–recall analysis. We considered the haplotypes inferred in all windows by the clustering algorithm and by the cut-off method based on the minimum number of reads supporting the variant. Red circles represent precision and recall for a set of threshold values chosen in the cut-off method (values from 1 to the number of reads in the most-supported haplotype), red arrows annotate points for a cut-off equal to 50. We performed a similar analysis on the output of the clustering algorithm, considering haplotypes whose confidence value (posterior probability) was greater or equal than a given threshold. Blue squares represent threshold values from 0.01 to 1, with blue arrows annotating the values obtained when the threshold is 0.9. Dashed lines and arrows are used for points obtained in the non-PCR-ampified sample, solid lines and arrows for points in the PCR-amplified one. In the non-PCR-amplified sample, we have a perfect precision (no false positives), and very good results for the recall. In the PCR-amplified sample, some false positives are found. In both cases, the performance of the clustering method is superior to the cut-off method. Results for individual windows can be found in Supplementary Data.
These results are confirmed in each individual window of the MSA (Supplementary Figures 7 and 8). The local analysis also highlights the fact that for the cut-off method, there is no single optimal cut-off value. In contrast, the posterior haplotype probability is interpretable across experiments and the default choice of 0.9, which we fixed for all further analyses, results in good performance throughout (Figure 2, blue arrows).
Error correction
The non-PCR-amplified sample showed a per-base error rate of 0.05%, while the amplified sample had a higher error rate of 0.25%. As a consequence, ~11% of the reads in the non-PCR sample and 43% in the PCR sample have one or more sequencing errors. After the error correction procedure, the error rate drops to 0.03% and 0.05% for the non-PCR and PCR sample, respectively, which results in ~92% of the reads being error free in both samples.
Frequency estimation
We assessed the ability of the ShoRAH algorithm to estimate the frequency of individual clones in the population. For the non-PCR-amplified sample, almost all haplotypes were reconstructed correctly and their frequency estimates were highly correlated with the ground truth (Pearson's correlation coefficient 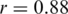 for all haplotypes and
for all haplotypes and 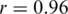 if outliers, i.e. haplotypes with
if outliers, i.e. haplotypes with 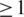 mismatches, are excluded; Figure 3a). Both haplotype reconstruction and frequency estimation are more difficult for the PCR-amplified sample, as shown by an increased number of imperfect haplotype matches and more discrepant frequency estimates (
mismatches, are excluded; Figure 3a). Both haplotype reconstruction and frequency estimation are more difficult for the PCR-amplified sample, as shown by an increased number of imperfect haplotype matches and more discrepant frequency estimates (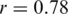 for all and
for all and 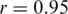 for all perfect matching haplotypes; Figure 3b).
for all perfect matching haplotypes; Figure 3b).

Frequency estimation with the clustering method. In each window true frequency of the haplotypes was estimated by aligning the raw reads to the original sequences (direct mapping) for non-PCR-amplified sample (a) and PCR-amplified sample (b). Then, haplotypes were reconstructed and it was checked whether they matched the originals in identity and frequency. Circles represent perfect matches with one of the original haplotypes, triangles indicate imperfect match. Except for a few spurious cases at low frequencies, there is good agreement both in identity and frequency between inferred and actual haplotypes.
Perfect reconstruction and frequency estimation was possible for many haplotypes with frequencies as low as 1% for the non-PCR sample and 0.1% for the PCR sample. This difference in resolution can be explained by the different average coverage of ~2100 and 6000base pairs per sequence position for the non-PCR and PCR sample, respectively. In each window, the sum of the frequencies of all true haplotypes that we were able to detect was always >96.5% for the non-PCR sample and >99.0% for the PCR sample.
Clinical samples
We applied error correction, haplotype reconstruction and frequency estimation to NGS data obtained from two clinical samples derived from two HIV-infected patients suspected to be in the same infection chain. The ShoRAH algorithm was run on the aligned reads and the output was used to make inference about the genetic diversity of the two viral quasispecies and about the distribution of drug resistance among individual clones of the populations. We focused on the 99 codons of the HIV-1 protease and considered all reads that covered this 297bp region completely: 891 reads for Patient 1 and 3272 reads for Patient 2.
In Figure 4, the allele frequency spectrum is shown after translation into amino acids. The consensus sequences for Patients 1 and 2 share the following mutations relative to the HXB2 reference strain: V3I, T12S, L19I, S37N, I54V, D60E, L63P, V77I and I93L, some of which are associated with resistance to PIs (16). They differ only at Position 10, where for Patient 1 an isoleucine is observed and a valine for Patient 2. However, we identified several additional mutations at low frequencies and the pattern of this variation was very different between the two patients. Sanger sequencing would not be able to detect this variation, because the frequency of most mutations is far below it detection limit of ~20% (Figure 4).

HIV protease amino acid allele frequency spectra of two patient samples. We analysed the frequency of amino acid substitution in the protease for two patients suspected to be in the same infection chain. They both present the drug resistance mutation I54V. Their consensus sequences differ at Position 10, Patient 1 showing an isoleucine and Patient 2 a valine. The horizontal line shows the 20% threshold typical of Sanger sequencing.
The allele frequency spectrum provides only a summary of the underlying population structure that ignores covariation, or phasing, of mutations at different sites of the genome. The real strength of NGS is to locally resolve the population structure. On the DNA level, we identified a total of 13 different haplotypes for Patient 1, and 15 different haplotypes for Patient 2, all with posterior probabilities close to one, which gave rise to five and six different protein sequences for Patients 1 and 2, respectively, (Supplementary Table S1). Using the cut-off method and a threshold of 50 observations to support a variant, we would call only 2 haplotypes for Patient 1 and 12 for Patient 2.
To compare the diversity of the two protease quasispecies, we employed two statistical tests on the estimated virus populations, i.e. on the inferred haplotype sequences and frequencies. A general non-parametric procedure for comparing vectors with categorical components was used to detect differences between the two haplotype distributions (36). The permutation test indicated that the observed difference between the quasispecies could not be explained by the sampling variance alone (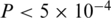 ) and hence is more likely to be the result of evolutionary change. We also quantified the amount of genetic diversity found in either population using the Simpson's index (37). The Simpson's, index is defined as
) and hence is more likely to be the result of evolutionary change. We also quantified the amount of genetic diversity found in either population using the Simpson's index (37). The Simpson's, index is defined as 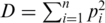 , where
, where 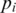 is the frequency of the
is the frequency of the 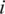 -th haplotype. It is the probability that two sequences that are randomly drawn from the population are identical. Thus, a low Simpson's, index indicates a high level of diversity. We found significantly higher viral genetic diversity in Patient 1 (
-th haplotype. It is the probability that two sequences that are randomly drawn from the population are identical. Thus, a low Simpson's, index indicates a high level of diversity. We found significantly higher viral genetic diversity in Patient 1 (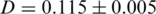 ) than in Patient 2 (
) than in Patient 2 (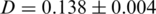 ).
).
The reconstructed virus populations show a diverse pattern of drug resistance mutations. PI resistance can be caused by many different protease mutations and usually it is mediated by combinations of these (27). To account for the effect of mutational patterns, we predicted, for each inferred haplotype, the level of phenotypic drug resistance to the PIs atazanavir, lopinavir, amprenavir, saquinavir and tipranavir (see ‘Materials and Methods’ section). The results are reported in Supplementary Table S1 and visualized for lopinavir in Figure 5 and for all five drugs in Supplementary Figure S9. Haplotype sequences are shown as discs in the plane in such a way that their pairwise distances reflect the Hamming distances in sequence space. The size of the discs is proportional to the haplotype frequency and the colour indicates the level of resistance. This analysis revealed variation in the predicted levels of PI resistance not only between the two patient samples, but also within individual quasispecies. For example, the viral quasispecies of Patient 1 displays differential levels of resistance to four of the five PIs, and the virus population of Patient 2 contains a low-frequency clone with reduced susceptibility to lopinavir and tipranavir. The latter haplotype would almost certainly go undetected with Sanger sequencing.

Structure of the viral quasispecies and predicted resistance to lopinavir. Circles represent detected haplotypes translated into amino acid sequences. The size reflects the frequency of the amino acid sequences, while the fill colour indicates the predicted resistance to the PI lopinavir. Green indicates higher and red lower levels of predicted drug susceptibility. The circles are positioned in the plot such that their distance approximately preserves the Hamming distance of the amino acid sequences. The number and the letter next to each circle denote, respectively, the patient and the protease sequence reported in the Supplementary Data.
DISCUSSION
We have shown how NGS can detect low-frequency variants in a mixed sample, provided that the technical sequencing errors are properly treated. Without this step, technical artefacts can easily be mistaken for real variants, and the amount of variation detected would necessarily overestimate the real one. In the case of HIV infection, low-frequency drug resistance mutations have been correlated with treatment failure (14,15). Thus, reliable estimates of these minority variants are crucial in effectively tailoring anti-viral therapy.
Different error sources can affect the reliability of these estimates. Reverse transcription and PCR amplification can introduce mismatches in the DNA sequences. Recombination between different templates in PCR can introduce haplotypes not existing in the original sample, and haplotypes can be amplified with different efficiency in the PCR reaction, altering the frequency spectrum of the original mixture. Finally, the sequencing process is far from being error-free.
We designed control experiments in order to investigate several error sources in a quantitative manner. In particular, we assessed two types of errors introduced by PCR amplification: recombination and amplification bias. We found that a fraction of <2% of the reads most likely originated from cross-over events during PCR amplification. Selective amplification (a change in the spectrum of frequencies after PCR) was also observed (see Control experiments in ‘Materials and Methods’ section). We conclude that PCR reactions must be designed and performed with great care in order to avoid artificial recombinants and to decrease the amount of bias in the investigated sample (33).
However, in our analysis we could separate these error sources from those due to pyrosequencing, and we could assess the performance of the ShoRAH algorithm used to correct sequencing errors. We obtained up to a five-fold decrease in the error rate, from 0.25% to 0.05% per base.
The cut-off method often used to detect minority variants requires previous knowledge of the sequencing error rate in order to set an optimal threshold for the minimum number of reads to call a variant. In contrast, the Bayesian method employed here does not require the error rate as input, but estimates it from the data. Using data from the control experiments we showed that, in fact, any choice of such a cut-off value results in poor haplotype reconstructions suffering from either low precision or low recall, and often both.
We stress that, unlike conventional Sanger sequencing, deep sequencing can detect the co-occurrence of mutations at least at a distance of the read length. Full HIV protease haplotypes were reconstructed from samples obtained from infected patients and showed how in a single host a diverse pattern of drug resistance can be observed. In HIV as well as in other viral infections, it will be of interest to investigate whether therapeutic interventions will benefit from the higher level of detail at which the viral population can be studied.
The data present in drug resistance databases, such as those used for the predictions shown in Figure 5, report phenotypic resistance to antiviral drugs of the entire population, but the matched DNA sequences are obtained by traditional Sanger sequencing. These data cannot inform about the contribution of individual clones to the level of resistance of the whole population. Therefore, the predictions shown in Figure 5 should be interpreted as the level of phenotypic drug resistance that can be expected in case the respective clone becomes dominant in the population. From a therapeutic point of view, the structure of the whole population is likely to have prognostic value, because pre-existing resistant minority clones will be selected rapidly as soon as the corresponding selective drug pressure is applied. In the future, models for predicting phenotypic drug resistance might include information on the viral population structure rather than the consensus sequence alone, eventually leading to a better understanding of resistance mechanisms.
Reliable reconstruction of the virus population is a prerequisite for making inference about the population structure, and it can serve as a starting point for epidemiological and clinical investigations. For example, the pattern of diversity observed in Figure 5 shows that the intra-patient diversity is larger than the inter-patient diversity, suggesting a close epidemiological link. Although it is in general difficult to establish the occurrence and the direction of transmission events with certainty, reliable high-resolution estimates of the population structure can be used in phylogenetic analyses to support or reject transmission hypotheses (38).
The knowledge of the population structure at the local level (on the order of the read length) can be used to reconstruct the haplotypes at a global level. Combinatorial approaches that assemble the reads after error correction into longer haplotypes have been proposed (25). Together with information obtained from paired-end reads, local structure estimates impose constraints on the global structure of haplotypes. We envisage extending our model of read generation from diverse haplotypes to the reconstruction of larger regions, and even the entire viral genome in the future.
An important step will be extending the ShoRAH algorithm to read data obtained from other NGS technologies, including the Illumina and SOLiD platforms, and highlighting the advantages and disadvantages of each in this specific application. Moreover, we foresee a better usage of base-specific quality scores, which we used here only to discard low-quality reads. The simple error model used in our approach does not distinguish between different substitutions as it considers the probability of a technical error independent of both the real and the erroneous base. It has been observed, for example for the Illumina platform (39), that some substitutions occur more frequently than others, and a model with additional parameters might take this into account. Such a model might be more accurate, but would not be efficient in correcting technical insertions and deletions. For this reason, we believe that another important contribution might be to perform alignment and error correction within the same probabilistic framework. A promising effort in this direction, though directed to metagenomics, has been recently presented (40).
Genetic diversity is important not only in retrovirus infections, but also in cancer and bacterial communities (41). We imagine the application of error correction to NGS data to all cases where sequencing is targeted towards a genetically heterogeneous sample and the goal is to estimate the structure of the population. Understanding the relation between phenotypic features of a pathogen population and its genetic structure will help in understanding the pathogenicity of parasite populations and possibly lead to the discovery of novel therapeutic options.
FUNDING
Swiss National Science Foundation (grant number CR32I2_127017) (in part).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
Patrick Knupfer is kindly acknowledged for calculating the predicted drug resistance to antiviral drugs of the clinical samples.
REFERENCES
Articles from Nucleic Acids Research are provided here courtesy of Oxford University Press
Full text links
Read article at publisher's site: https://doi.org/10.1093/nar/gkq655
Read article for free, from open access legal sources, via Unpaywall:
https://academic.oup.com/nar/article-pdf/38/21/7400/16771505/gkq655.pdf

Free to read at nar.oxfordjournals.org
http://nar.oxfordjournals.org/cgi/content/full/38/21/7400
Free to read at nar.oxfordjournals.org
http://nar.oxfordjournals.org/cgi/reprint/38/21/7400.pdf
Free to read at nar.oxfordjournals.org
http://nar.oxfordjournals.org/cgi/content/abstract/38/21/7400
Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Article citations
Next-Generation Sequencing Reveals a High Frequency of HIV-1 Minority Variants and an Expanded Drug Resistance Profile among Individuals on First-Line ART.
Viruses, 16(9):1454, 13 Sep 2024
Cited by: 0 articles | PMID: 39339930 | PMCID: PMC11437406
A study on factors influencing delayed sputum conversion in newly diagnosed pulmonary tuberculosis based on bacteriology and genomics.
Sci Rep, 14(1):18550, 09 Aug 2024
Cited by: 0 articles | PMID: 39122761 | PMCID: PMC11315884
Methods to improve the accuracy of next-generation sequencing.
Front Bioeng Biotechnol, 11:982111, 20 Jan 2023
Cited by: 10 articles | PMID: 36741756 | PMCID: PMC9895957
Review
 Free full text in Europe PMC
Free full text in Europe PMCK-Mer Spectrum-Based Error Correction Algorithm for Next-Generation Sequencing Data.
Comput Intell Neurosci, 2022:8077664, 14 Jul 2022
Cited by: 1 article | PMID: 35875730 | PMCID: PMC9303089
Detection of Chimeric Cellular: HIV mRNAs Generated Through Aberrant Splicing in HIV-1 Latently Infected Resting CD4+ T Cells.
Front Cell Infect Microbiol, 12:855290, 28 Apr 2022
Cited by: 2 articles | PMID: 35573784 | PMCID: PMC9096486
Go to all (136) article citations
Data
Data behind the article
This data has been text mined from the article, or deposited into data resources.
BioStudies: supplemental material and supporting data
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
Analysis of 454 sequencing error rate, error sources, and artifact recombination for detection of Low-frequency drug resistance mutations in HIV-1 DNA.
Retrovirology, 10:18, 13 Feb 2013
Cited by: 71 articles | PMID: 23402264 | PMCID: PMC3599717
HIV drug resistance surveillance using pooled pyrosequencing.
PLoS One, 5(2):e9263, 17 Feb 2010
Cited by: 21 articles | PMID: 20174661 | PMCID: PMC2822863
Sequencing-based detection of low-frequency human immunodeficiency virus type 1 drug-resistant mutants by an RNA/DNA heteroduplex generator-tracking assay.
J Virol, 78(13):7112-7123, 01 Jul 2004
Cited by: 31 articles | PMID: 15194787 | PMCID: PMC421662
Error rates, PCR recombination, and sampling depth in HIV-1 whole genome deep sequencing.
Virus Res, 239:106-114, 27 Dec 2016
Cited by: 30 articles | PMID: 28039047
Review
Funding
Funders who supported this work.
Swiss National Science Foundation (1)
Grant ID: 127017


