The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking.
![]() Free full text in Europe PMC
Free full text in Europe PMC
Abstract
Free full text
The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking
Associated Data
Abstract
Ciliopathies are a genetically and phenotypically heterogeneous group of human developmental disorders whose root cause is the absence or dysfunction of primary cilia. Joubert syndrome is characterized by a distinctive hindbrain malformation variably associated with retinal dystrophy and cystic kidney disease. Mutations in CC2D2A are found in ~10% of patients with Joubert syndrome. Here we describe the retinal phenotype of cc2d2a mutant zebrafish consisting of disorganized rod and cone photoreceptor outer segments resulting in abnormal visual function as measured by electroretinogram. Our analysis reveals trafficking defects in mutant photoreceptors affecting transmembrane outer segment proteins (opsins) and striking accumulation of vesicles, suggesting a role for Cc2d2a in vesicle trafficking and fusion. This is further supported by mislocalization of Rab8, a key regulator of opsin carrier vesicle trafficking, in cc2d2a mutant photoreceptors and by enhancement of the cc2d2a retinal and kidney phenotypes with partial knockdown of rab8. We demonstrate that Cc2d2a localizes to the connecting cilium in photoreceptors and to the transition zone in other ciliated cell types and that cilia are present in these cells in cc2d2a mutants, arguing against a primary function for Cc2d2a in ciliogenesis. Our data support a model where Cc2d2a, localized at the photoreceptor connecting cilium/transition zone, facilitates protein transport through a role in Rab8-dependent vesicle trafficking and fusion.
INTRODUCTION
Ciliopathies are a recently defined and rapidly growing group of human disorders caused by dysfunction of primary cilia which are microtubule-based organelles present on the surface of most differentiated cell types (1–3). Despite significant functional differences in diverse cell types, all primary cilia share a common basic structure consisting of a circular array of nine microtubule doublets forming the axoneme, which is anchored in the basal body, a modified centriole (4–6). Dysfunction of primary cilia results in a spectrum of phenotypes including central nervous system malformations, retinal dystrophy and cystic kidney disease as illustrated in Bardet–Biedl syndrome (MIM 209900), Joubert syndrome (MIM 213300) or Senior-Løken syndrome (MIM 266900), among others (7). Zebrafish models for ciliopathies [sco/arl13b, ift88 and ift57 mutants (8–11); cep290, bbs and nphp gene morphants (12–16)] recapitulate the cystic kidney and retinal phenotypes and offer insight into the mechanisms underlying these defects (17,18).
The photoreceptor outer segment is a specialized primary cilium consisting of a microtubule-based axoneme and regular stacks of membrane folds or disks containing the photopigments (opsins) required for the phototransduction cascade (17,19). Development/maintenance of the outer segment requires transport of at least some of its components by complex networks of proteins involved in: (i) vesicle sorting and trafficking from the Golgi to the base of the cilium, (ii) docking at the base of the cilium and fusion with the periciliary membrane and (iii) transport within the ciliary shaft dependent on intra-flagellar transport (IFT) (19,20). The vesicle trafficking steps are controlled by several small GTPases from the Rab family among which Rab8 plays a central role in fusion of opsin-carrier vesicles at the base of the cilium (21). Roles for Rab8 in photoreceptor outer segment disk morphogenesis (22) and ciliary membrane biogenesis in cooperation with the BBSome (23) have also been shown.
Joubert syndrome (MIM 213300) is an autosomal recessive human ciliopathy characterized by cerebellar vermis hypoplasia associated with variable involvement of other organ systems, including retinal dystrophy and cystic kidney disease (24–27). Significant genetic heterogeneity underlies Joubert syndrome with 11 causative genes identified to date: CC2D2A, RPGRIP1L, NPHP1, AHI1, CEP290, TMEM67 (MKS3), ARL13B, INPP5E, OFD1, TMEM216 and TCTN2 (12,28–38). Their products localize to the basal body, transition zone (39–42) and/or ciliary axoneme (31,32) and are involved in a number of ciliary-related functions, including ciliogenesis [ARL13b (11), TMEM216 (40), AHI1 (42) or CEP290 (43)] and regulation of ciliary protein content [ARL13B (44), RPGRIP1L (41), AHI1 (45), CEP290 (39), MKS3 (41)]. In addition, a role in vesicle trafficking has been suggested for AHI1 and CEP290 based on their interaction with Rab8 (42,43). Mice deficient for these genes display abnormal retinal photoreceptor outer segments and opsin mislocalization (45–47).
The Joubert-associated protein CC2D2A localizes to the base of the cilium in cultured retinal pigment epithelium (RPE) and kidney (IMCD3) cells (30), and contains a phospholipid-binding C2 domain found in proteins involved in vesicle fusion and trafficking (48), as well as coiled-coil domains found in many ciliary proteins. Tallila et al. (49) reported absence of cilia in cultured human fibroblasts homozygous for a CC2D2A mutation and therefore proposed that CC2D2A is required for cilium formation. More recent work using Caenorhabditis elegans localized CC2D2A protein to the transition zone of amphid and phasmid neuron cilia and found no structural cilium defects in cc2d2a-mutant worms (41). This study proposed that CC2D2A functions in a complex with other ciliopathy-associated proteins at the transition zone of cilia as a ‘gate-keeper’, restricting proteins from inappropriately entering the cilium. This function is similar to what has been described for CEP290 in a Chlamydomonas model (39).
We previously reported a zebrafish cc2d2a mutant (snl/cc2d2aw38), harboring a nonsense mutation up-stream of the C2 domain (W628X), that displays a cystic kidney phenotype (30,50). This phenotype was enhanced by partial knock-down of cep290, indicating a genetic interaction between cc2d2a and cep290. Furthermore, a physical interaction between the products of these genes was demonstrated (30). In this new work, we explore the cell-biological role of Cc2d2a in zebrafish photoreceptors. We show that Cc2d2a localizes to the connecting cilium/transition zone of cilia in photoreceptors and various other cells and is required for outer segment development and normal visual function. Our analysis reveals mislocalization of transmembrane outer segment proteins (opsins) and striking accumulation of vesicles in mutant photoreceptors, suggesting a role for Cc2d2a in vesicle trafficking and fusion. This is further supported by mislocalization of Rab8 in cc2d2a−/− photoreceptors and by a synergistic effect between cc2d2a and rab8 in vivo. We also demonstrate that cilia are present in photoreceptors and other cell types where cc2d2a is normally expressed, arguing against a primary function for Cc2d2a in ciliogenesis. Our data support a model where Cc2d2a, localized at the connecting cilium of the photoreceptor, functions in Rab8-dependent trafficking and fusion of opsin-carrier vesicles.
RESULTS
cc2d2a is required for zebrafish photoreceptor outer segment development
Previous phenotypic analysis of the cc2d2aw38 zebrafish mutant (hereafter referred to as cc2d2a mutant or cc2d2a−/−) revealed a sinusoidal body shape in larvae and adults (50) (Supplementary Material, Fig. S1A and B) and an incompletely penetrant pronephric (kidney) cyst phenotype with 30–50% of homozygous mutants developing cysts by 3 days post-fertilization (d.p.f.) (30). Given the retinal dystrophy seen in human patients with CC2D2A-associated Joubert syndrome (30,51), we evaluated the eyes of homozygous cc2d2a zebrafish mutants (cc2d2a+/− embryos were indistinguishable from wild-type embryos in all assays). While overall eye size and retinal lamination are unaffected on light microscopy (Fig. 1A and B and Supplementary Material, Fig. S1C and D), rod and cone outer segments are short and disorganized in cc2d2a−/− fish compared with wild-type and heterozygous siblings at 5 d.p.f. (red brackets in Fig. 1A–D). This phenotype is variable even within a single-mutant retina, ranging from shortened but otherwise normally shaped outer segments to complete absence of outer segments. Intermediate severity is most commonly observed, with disorganized whorls of membrane visible above the mitochondria in the inner segment of the photoreceptors (red arrow in Fig. 1D and F). Apical–basal polarity is unaffected based on preservation of the characteristic photoreceptor cell body shape, highlighted with the zpr1 antibody which marks red/green cones (52) (Fig. 1J′), as well as the normal distribution of organelles as viewed by transmission electron microscopy (Fig. 1D).

cc2d2a is required for zebrafish photoreceptor outer segment development. (A and B) Plastic sections of 5 d.p.f. cc2d2a+/+ (A) and cc2d2a−/− (B) retinas. Black brackets highlight the photoreceptor cell layer, and red brackets highlight the outer segments. (C–F) Transmission electron microscopy on 5 d.p.f. retinal sections shows disorganized stacks of membrane (red arrow) in the outer segments of cc2d2a−/− retinas (D) compared with wild-type (C). Higher power views (E and F) show whorls of membrane stacks replacing the outer segment (red arrow). (G and H) Tg(TaCP:MCFP) expression in 80 h.p.f. cryosectioned retinas. Wild-type cc2d2a+/+ or +/− cone photoreceptors have readily visible outer segments (G), whereas cc2d2a−/− photoreceptors have only short outer segments without the typical shape at the same stage (H). (I–J′) Red–green cones labeled with zpr1 antibody in wild-type (I and I′) and cc2d2a−/− (J and J′) 5d.p.f. retinal cryosections showing comparable density of cone photoreceptors and grossly preserved photoreceptor cell morphology except in the apical segment. (K and L) Red–green cone cell bodies labeled with zpr1 antibody (red) in retinal cryosections from 4-week-old wild-type (K) and cc2d2a−/− fish (L). Fluorescent images are single-confocal sections. M, mitochondria; N, nuclei; RP, retinal pigment. Scale bars are 10 µm in (A and B), 2 µm in (C and D), 0.5 µm in (E and F), 4 µm in (G and H), 20 µm in (I and J) and 10 µm in (K and L).
To determine whether outer segments initially form normally and subsequently degenerate in cc2d2a mutants, we used the tg(TaCP:MCFP) line, in which membrane-tagged CFP is expressed under the transducin promoter specifically in cone photoreceptors, thereby highlighting all cone outer segments (53). MCFP expression is robustly detected at 80 h post-fertilization (h.p.f.) in cryosections from wild-type embryos demonstrating well-organized nascent cone outer segments (Fig. 1G). In cc2d2a−/− retinas, few outer segments are visible, and these appear short and disorganized even at this early stage, indicating that outer segment formation is never achieved normally (Fig. 1H). Other zebrafish mutants with abnormal or absent photoreceptor outer segments such as ift88, ift57, fleer and elipsa all display significant photoreceptor cell loss, predominantly in the central retina, by 5d.p.f. (8,54). In contrast, cc2d2a−/− embryos have relatively normal numbers of photoreceptors at this stage as shown by immunostaining of cryosections with zpr1, which marks red–green cones (52) (Fig. 1I and J), and 4c12, which marks rods (55) (Supplementary Material, Fig. S1G and H). In addition, we did not observe increased cell death based on caspase staining at 4 d.p.f. (Supplementary Material, Fig. S1F). In contrast, retinas from 4-week-old cc2d2a−/− fish show moderate loss of photoreceptors, as shown by zpr1 (Fig. 1K and L) and 4c12 (Supplementary Material, Fig. S1I and J) staining. This loss of photoreceptors is even more clearly visible using bodipy counterstaining (Supplementary Material, Fig. S1K and L) which highlights cell membranes and membrane-rich outer segments, thereby indicating that photoreceptor cell loss does occur in cc2d2a mutants, but at a slower rate than in other ciliopathy mutants. Based on these data, we conclude that Cc2d2a is required for photoreceptor outer segment development and maintenance, and that its deficiency leads to slow photoreceptor degeneration.
Cc2cd2a is required for normal visual function
Given the retinal photoreceptor phenotype observed in homozygous cc2d2a mutants, we evaluated the mutant fish for abnormal visual function. A small proportion of homozygous cc2d2a mutants are viable to adulthood, and although small and morphologically highly abnormal (Supplementary Material, Fig. S1A and B), they display some visual function based on relatively normal feeding behavior and responsiveness to objects moving in front of the tank. We performed optokinetic response testing (56,57) in 5d.p.f. larvae and found no obvious difference in unquantified eye movements between mutants and their wild-type or heterozygote siblings at high-contrast conditions (data not shown). However, electroretinogram (ERG) recordings (58) at 5 and 6d.p.f., using a series of light intensities, revealed significantly decreased responses in mutants compared with wild-type and heterozygous siblings at all tested conditions (Fig. 2). Therefore, the observed retinal photoreceptor outer segment phenotype is associated with decreased visual sensitivity in cc2d2a mutant zebrafish.

Visual function is abnormal in cc2d2a−/− zebrafish. (A) Representative ERG from cc2d2a+/+ or +/− 6 d.p.f. fish (n = 6). (B and C) ERGs from mildly (B) and severely (C) affected cc2d2a−/− 6 d.p.f. fish (n = 6). Different tracings in each graph represent different light intensities.
Cc2d2a localizes to the connecting cilium/transition zone of cilia in photoreceptors and various other cells
The products of Joubert/ciliopathy spectrum genes localize to specific segments of the cilium, ranging from the basal body [AHI1 (42), for example] to the ciliary axoneme [ARL13B (32) or INPP5E (31)], and their precise localization can be informative with respect to their function in ciliary biology. We previously showed that CC2D2A co-localizes at the base of cilia with CEP290 in cultured RPE cells (30). In addition, a recent work has shown that the C. elegans CC2D2A ortholog, along with a subset of other ciliopathy proteins, localizes to the transition zone (41), a region at the base of the cilium just apical to the basal body. To determine whether this subcellular localization is conserved in photoreceptors and other ciliated cell types, we developed a monoclonal antibody against amino acids 430–1019 of the zebrafish protein. This antibody detects a protein of the expected 191 kDa molecular weight in lysates from 48 h.p.f. wild-type embryos but not in lysates from embryos lacking Cc2d2a (Fig. 3I), indicating lack of full-length Cc2d2a protein in cc2d2aw38 mutants. Moreover, the antibody recognizes the truncated Cc2d2a protein generated by the W628X mutation when overexpressed in HEK293T cells (Fig. 3J). This band is absent in lysates from cc2d2a mutants, thereby indicating that no truncated Cc2d2a protein is present and that the cc2d2aw38 mutant used in this study is a protein null.

Cc2d2a localizes to the connecting cilium/transition zone of cilia in photoreceptors and other cell types. (A–C′″) Retinal cryosections from cc2d2a+/+ embryos labeled with anti-Cc2d2a (green) and anti-acetylated tubulin [red in (A–A′″)], anti-Ift88 [red in (B–B′″)] and anti-gamma-tubulin [red in (C–C′″)] show Cc2d2a localization at the base of cilia (arrows). The Cc2d2a signal is apical to the basal-most acetylated tubulin signal [arrowhead in (A″)], apical to the gamma-tubulin signal (C–C′″) and within the Ift88-poor region that represents the connecting cilium, between the brightest Ift88 signal and a fainter, more basal signal [arrowheads in (B′ and B″)]. Embryos are 4 d.p.f. in (A) and 5 d.p.f. in (B and C). (D–G) Immunofluorescence against Cc2d2a (green) and Arl13b (red) showing Cc2d2a localization at the base of 48 h.p.f. pronephric duct cilia (D and D′), 24 h.p.f. floorplate cilia (E and E′), 72 h.p.f. olfactory pit cilia (F) and 24 h.p.f. cerebellar neuronal progenitor cilia (G). (H) High power confocal image of a neuronal progenitor cilium from a 24 h.p.f. arl13b-GFP embryo stained with antibodies against gamma-tubulin (green), Cc2d2a (red) and GFP (blue). (D–H) Single-confocal sections of whole-mount embryos. Scale bars are 6 µm in (A), 2 µm in (A′), 0.4 µm in (A″ and A′″), 4 µm in (B and C), 1 µm in (B′–B′″), 0.5 µm in (C′–C′″), 5 µm in (D and F), 10 µm in (E), 20 µm in (G) (2 µm in inset) and 0.6 µm in (H). (I) Western blot with Cc2d2a P2D9 antibody from whole wild-type, zygotic and maternal zygotic (mz) cc2d2a−/− embryo lysates at 48 h.p.f. The Cc2d2a band appears as a doublet around 190 kDa in wild-type (cc2d2a+/+) and is absent in zygotic and mz cc2d2a−/− embryos. (J) Western blot with Cc2d2a P2D9 antibody on full-length and truncated (W628*) Cc2d2a-GFP protein overexpressed in HEK293T cells. The antibody recognizes the full-length as well as the truncated Cc2d2a protein.
Immunostaining on retinal cryosections at 4 and 5 d.p.f. confirms Cc2d2a localization at the base of acetylated tubulin- and Ift88-stained photoreceptor cilia (Fig. 3A–B′), just apical to gamma-tubulin staining (Fig. 3C and C′). Moreover, higher resolution images confirm Cc2d2a localization at the connecting cilium of zebrafish photoreceptors, since the Cc2d2a signal is (i) slightly apical to the basal-most acetylated tubulin staining (arrowhead in Fig. 3A″), (ii) just apical to gamma-tubulin staining of the basal body (Fig. 3C′) and (iii) contained in the Ift88-poor region previously shown to define the connecting cilium in photoreceptors (arrow in Fig. 3B′–B′″) (59). We used whole-mount embryos to assess cilia in other tissues and found that Cc2d2a is localized at the base of a variety of Arl13b-stained cilia, including motile cilia in 48 h.p.f. pronephric ducts (Fig. 3D and D′) and 24h.p.f. floorplate cells (Fig. 3E and E′), sensory cilia of 3 d.p.f. olfactory neurons (Fig. 3F) and primary cilia of 24 h.p.f. neuronal progenitors in the cerebellum (Fig. 3G and inset). This staining is entirely absent in cc2d2a−/− embryos (Supplementary Material, Fig. S2A–F), confirming the specificity of the antibody. Cc2d2a also localizes to the transition zone in neuronal progenitor cilia, where the Cc2d2a signal is detected between the gamma-tubulin basal body signal and the Arl13b axonemal signal (Fig. 3H), partially overlapping with each, as confirmed by fluorescence intensity graphs (Supplementary Material, Fig. S2G). Our previous description that CC2D2A and CEP290 co-localize in cultured cells further supports this finding, since CEP290 has also been shown to localize to the transition zone (39). Together, these data confirm localization of Cc2d2a to the connecting cilium in photoreceptors and to the transition zone of primary and motile cilia in a variety of cell types.
Cc2d2a is not essential for cilia assembly
The products of other Joubert syndrome genes such as CEP290, AHI1, ARL13b or RPGRIP1L are thought to control ciliogenesis based on the observations that (i) cultured cells treated with siRNA/shRNA against CEP290 (43) or AHI1 (42) fail to extend cilia; (ii) mouse embryonic fibroblasts deficient for AHI1 have reduced numbers of cilia (42); and (iii) cilia are abnormal in zebrafish and mice deficient for Arl13b (11,60) and Rpgrip1l (61). In addition, Tallila et al. (49) reported that human fibroblasts with a truncating mutation in CC2D2A also lack cilia, supporting a role for CC2D2A in cilia formation. Since photoreceptor outer segments are highly specialized primary cilia, a defect in ciliogenesis could explain the outer segment phenotype observed in cc2d2a−/− fish. To test this hypothesis, we sought to determine whether cilia form normally in cc2d2a−/− retinas. Despite defective outer segment development in mutant photoreceptors, we observe structurally normal connecting cilia positioned apically within the inner segment using transmission electron microscopy (brackets in Fig. 4A and B, arrow in Fig. 6C and D). Furthermore, the total number of cilia is not decreased in mutant retinas as shown by acetylated tubulin immunostaining on whole-mount eyes from 5 d.p.f. Tg (TaCP: MCFP) cc2d2a−/− larvae (apical views in Fig. 4C–F): cc2d2a−/− eyes contain on average 13.1 ± 2.05 cilia/100 µm2 (n = 10 eyes) compared with 13.5 ± 1.6 cilia/100 µm2 in cc2d2a+/+ eyes (n = 5 eyes), P = 0.74 (Student's t-test). However, despite the normal density of cilia in mutant retinas, cilia length appeared decreased in mutants at the periphery of these whole-mount eyes, where the cilia can be viewed tangentially (Fig. 4E and F). To quantify the difference between wild-type and mutant axonemes, we measured the length of acetylated tubulin staining in equivalent retinal sections from multiple wild-type and mutant embryos at 7 d.p.f. (brackets in Fig. 4G and H). The average length of acetylated tubulin staining was 1.93 ± 0.87 µm in cc2d2a−/− photoreceptors compared with 3.8 ± 0.93 µm in cc2d2a+/+ or +/− photoreceptors (P < 0.0001, Student's t-test, n = 180 photoreceptors from 16 eyes). Although the length of the axoneme varies between different photoreceptor types (i.e. between cones and rods) and although some axonemes may leave the plane of the section, the results were very consistent across embryos and closely matched our observations in whole mounts.

cc2d2a is not required for ciliogenesis. (A and B) Connecting cilia (brackets) in wild-type (A) and cc2d2a−/− (B) photoreceptors demonstrated by transmission electron microscopy. (C and D) Apical view of ciliary axonemes labeled with acetylated tubulin antibodies (red) in wild-type (C) and cc2d2a−/− (D) whole-mount eyes in which cone outer segments are labeled with tg(TaCP:MCFP). (E and F) Peripheral views of the same eyes as in (C and D) showing only the acetylated tubulin staining. Note the shorter axonemes in cc2d2a−/− eyes (F) compared with wild-type (E). (G and H) Ciliary axonemes labeled with acetylated tubulin (red) and Ift88 (green) antibodies in wild-type (G) and cc2d2a−/− (H) retinal cryosections. Brackets highlight axonemal length. Embryos are 5 d.p.f. in (A–F) and 7 d.p.f. in (G and H). (I–N) Immunofluorescence with Arl13b antibody (red) highlighting cilia and with gamma-tubulin antibody (green) highlighting basal bodies in 24 h.p.f. whole-mount mz cc2d2a+/− (I, K and M) and mz cc2d2a−/− (J, L and N) embryos showing retinal neuronal progenitor cilia (arrowheads in insets of I and J), cerebellar neuronal progenitors (arrowheads in insets of K and L) and floorplate cilia (M and N). (O and P) Pronephric duct cilia stained with acetylated tubulin (red) and gamma-tubulin (green) antibodies in 48 h.p.f. whole-mount mz cc2d2a+/− (O) and mz cc2d2a−/− (P) embryos. The anterior–posterior axis is indicated by the arrows. (Q and R) Acetylated tubulin antibody staining of olfactory pit cilia at 3 d.p.f. in whole-mount wild-type (Q) and cc2d2a−/− (R) embryos. (I–L) and (Q and R) Single-confocal sections. (C–F) and (M–P) Projected stacks of confocal sections. Scale bars are 500 nm in (A and B), 5 µm in (C and D), 10 µm in (E and F), 4 µm in (G and H), 2 µm in (I–L), 10 µm in (M and N) and 5 µm in (O–R).

Loss of Cc2d2a leads to vesicle accumulation in photoreceptors. (A–H) Transmission electron microscopy images of 5 d.p.f. cc2d2a+/+ (A–A′ and G) and cc2d2a−/− (B–F and H) photoreceptors. (A) Low-power image of the inner segment and the base of the outer segment in cc2d2a+/+ photoreceptors showing the mitochondrial cluster M, the connecting cilium (arrow and white line) and the base of the outer segment (A′), with neatly stacked membranes [high power view (A′)]. (B) Low-power image of cc2d2a−/− photoreceptors with normal mitochondrial clusters M and nuclei N, but massive accumulation of vesicles around a normal-appearing connecting cilium [arrow and white line in (B)]. The black and white bracket highlights an outer segment replaced by vesicles. (B′) High-power image of the vesicles boxed in (B). (C and D) Higher power images of the accumulating vesicles (bracket) just below partially stacked membranes. (D) Cross-section through a connecting cilium (arrow) with nine normal appearing microtubule doublets. (E) Lower power image of a different region showing moderate vesicle accumulation (black bracket) below a recognizable outer segment (white bracket). Note the presence of vesicles lateral to the nucleus (red bracket). (F) Higher power image of the basal portion of a photoreceptor showing vesicle accumulations (bracket) between nuclei. (G and H) High-power images of the Golgi apparatus in wild-type (G) and cc2d2a−/− (H) photoreceptors. (I and J) Cryosections of transient transgenic cc2d2a+/+ (I) and cc2d2a−/− (J) embryos expressing the Golgi specific transgene tg(gnat2:Man2a)-RFP (red) under the transducin promoter control. Scale bars are 1 µm in (A–B), 100 nm in (A′ and B′), 200 nm in (C), 100 nm in (D), 2 µm in (E), 1 µm in (F), 500 nm in (G and H) and 2 µm in (I and J). M, mitochondria; N, nuclei; G, Golgi.
This does not reflect a general role for Cc2d2a in ciliogenesis or cilium extension, since many cell types that normally express Cc2d2a have normal-appearing cilia despite loss of Cc2d2a expression in cc2d2a mutants. Indeed, a variety of different cilia types are normal in cc2d2a−/− fish, including primary cilia in 24 h.p.f. retinal and neuronal progenitors (Fig. 4I–L, arrowheads in insets), as well as motile cilia in 24 h.p.f. floorplate (Fig. 4M and N) and 48 h.p.f. pronephric ducts (Fig. 4O and P). Similarly, olfactory cilia are indistinguishable between cc2d2a−/− and wild-type embryos at 3 d.p.f. (Fig. 4Q and R). For these experiments at early developmental stages (24 and 48 h.p.f.), we used embryos lacking both maternal and zygotic (mz) Cc2d2a to exclude the possibility that a mild phenotype at these earlier stages results from the perdurance of maternal gene product. While we cannot completely rule out a distinct role for Cc2d2a in photoreceptor axoneme elongation compared with other cell types, this possibility is unlikely based on the similar localization of Cc2d2a at the base of photoreceptors and other cilia, the presence of normal connecting cilia and normal-appearing axonemes in some mutant photoreceptors.
Cc2d2a is required for trafficking of transmembrane proteins to the outer segment
Proteins localized at the base of cilia can play a role in ciliogenesis but can also be involved in the transport of specific proteins to the ciliary membrane. This aspect of ciliary function has been particularly well studied in photoreceptors, since their outer segments are highly specialized primary cilia requiring massive transport of specific components (predominantly photopigments called opsins) (19). Opsins are transmembrane proteins that are normally targeted specifically to the outer segment by directed vesicle trafficking from the Golgi to the periciliary ridge complex, where fusion with the membrane occurs followed by IFT-dependent transport (20,62–64). Mutants in ciliopathy genes Cep290 and Ahi1 (45–47), as well as IFT components ift88, ift57 and Ift20 (8,65), exhibit mislocalization of opsins within the photoreceptor cell, suggesting a role for these proteins at one or more steps in the opsin transport.
We found a similar opsin mislocalization phenotype affecting both rod and cone photoreceptors in cc2d2a−/− fish. Immunostaining of 5 d.p.f. retinal cryosections with 4D2 (rhodopsin) and BOPS (blue cone opsin) antibodies showed that both rod and cone opsins are partially mislocalized throughout the photoreceptor cell (Fig. 5A–D). This is true not only in photoreceptors entirely lacking outer segments, but also in photoreceptors with grossly normal appearing, but shortened, outer segments. This trafficking defect does not affect all outer segment proteins, since transducin (66), a membrane-associated protein reaching the outer segment by diffusion (67,68), is not mislocalized in cc2d2a−/− photoreceptors (Fig. 5E and F). Moreover, trafficking towards other subcellular compartments occurs normally as well, as demonstrated by normal ribeye (69) localization to the synapse in cc2d2a−/− photoreceptors (Fig. 5G and H).

Selective trafficking defects in cc2d2a−/− photoreceptors. (A and B) BOPS localization (BOPS antibody, red) in wild-type (A) and cc2d2a−/− (B) photoreceptors. (C and D) Rhodopsin localization (4D2 antibody, red) in wild-type (C) and cc2d2a−/− (D) photoreceptors. Outer segments are indicated by brackets. Mislocalized photopigment (opsin) is indicated by white arrows. (E and F) Transducin (red) localization by immunofluorescence is restricted to the outer segments (indicated by brackets) in both cc2d2a+/+ and cc2d2a−/− photoreceptors. (G and H) Ribeye (red) localization at the synapse is indistinguishable between cc2d2a+/+ and cc2d2a−/− photoreceptors. (I–I″) BOPS staining [red in (I)] and Golgi expression of tg(gnat2:Man2a-RFP, green) do not significantly overlap [see arrowheads in (I) and (I″) for BOPS and arrow in (I) and (I′) for Golgi]. (J) Rhodopsin (4D2 antibody, red, arrow) is mislocalized between the nucleus (DAPI, blue) and the subcortical actin network stained with phalloidin (green, arrowhead). All images are single-confocal sections of 5 d.p.f. cryosections. Scale bars are 4 µm in all panels.
Additional information about the role of Cc2d2a in opsin trafficking can be gained by determining the subcellular localization of ectopic opsins. For example, a recent study of a conditional mouse Ift20 mutant found that mislocalized opsins are concentrated in the Golgi, and thereby suggested a role for Ift20 in coupling the transport of opsins through the endomembrane system to the IFT system for transport into the cilium (65). We expressed a fusion protein in cc2d2a−/− embryos that specifically localizes to the Golgi of cone photoreceptors (gnat2:Man2a-RFP) (70) and observed little co-localization with the mislocalized cone opsin (Fig. 5I–I″), indicating that Cc2d2a functions downstream of the Golgi apparatus in the pathway transporting opsins to the outer segment. This is consistent with Cc2d2a localization only at the base of cilia and not at the Golgi apparatus. In addition to the accumulation of opsins in the apical portion of the mutant photoreceptors (inner segment area), we also observe mislocalization of opsins all along the cell membrane and in the basal portion of the photoreceptors near the synapse (arrow in Fig. 5J). To determine whether the mislocalized opsins are contained within the cell membrane or whether they accumulate in the cytoplasm, we performed co-staining with rhodopsin antibody (red) and phalloidin (green) to mark subcortical actin. In the majority of cases, the mislocalized rhodopsin (arrow) is present between the DAPI-stained nuclei and the phalloidin-stained actin network (arrowhead), thereby indicating that the majority of the mislocalized opsins are not inserted in the plasma membrane but accumulate in the cytoplasm (Fig. 5J). Together, these data indicate that Cc2d2a functions in a post-Golgi step for opsin-carrier vesicle trafficking to the cilium.
Loss of Cc2d2a leads to vesicle accumulation in photoreceptors
To further evaluate a possible role for Cc2d2a in vesicle fusion, we examined 5 d.p.f. retinas using transmission electron microscopy and identified striking accumulations of vesicles in cc2d2a−/− eyes (Fig. 6B and B′, C–F). These vesicles are particularly abundant in the apical-most portion of the photoreceptors (the inner segment), with accumulations around the connecting cilium and apical to the mitochondria (Fig. 6B and B′). In some photoreceptors, these vesicles entirely replace the membrane stacks of the outer segment (Fig. 6B, black and white bracket), but in most photoreceptors, they accumulate below short, partially disorganized stacks of membranes (Fig. 6C and E, black brackets). In addition, vesicles are also located more basally, lateral to the nucleus (Fig. 6E, red bracket; 6F, black bracket). Electron microscopy studies performed on zebrafish mutants lacking outer segments such as IFT mutants ift88 and ift172 have not found similar accumulation of vesicles despite complete lack of outer segments (9), indicating that accumulation of vesicles is not simply a consequence of deficient outer segment formation. The initial steps in vesicle trafficking, namely budding from the Golgi apparatus, appear unaffected in the cc2d2a mutant, since we did not observe significant ultra-structural anomalies of the Golgi in cc2d2a−/− photoreceptors by transmission electron microscopy (Fig. 6H). In addition, we mosaically expressed the Golgi marker gnat2:Man2a-RFP (70) in cone photoreceptors, using the transducin promoter, and observed no difference in Golgi morphology between wild-type and cc2d2a−/− fish (Fig. 6I and J). These data provide ultra-structural evidence supporting a role for Cc2d2a in vesicle fusion at the base of the photoreceptor cilium, but not in the earlier steps of vesicle budding from the Golgi.
Cc2d2a is required for punctate Rab8 localization at the base of the outer segment
The small GTPase Rab8 is a key regulator of opsin-carrier vesicle trafficking in photoreceptors, based on the observation that a dominant-negative form of Rab8 leads to vesicle accumulation in Xenopus photoreceptors (21), similar to what we observe in cc2d2a mutants. This led us to evaluate whether Rab8 was normally localized in cc2d2a mutant photoreceptors. Since specific antibodies against zebrafish Rab8 are not available, we mosaically expressed a cherry-tagged Rab8 protein in cone photoreceptors using the transducin promoter. In cc2d2a+/+ and +/− photoreceptors, Rab8-cherry is concentrated in a single punctum at the base of the outer segment in 66% of expressing cells (Fig. 7A and A′ and Supplementary Material, Fig. S3E, quantification in Fig. 7C), consistent with a previous description for Rab8 localization in Xenopus photoreceptors using a Rab8-GFP construct (21). In the cc2d2a−/− siblings, this punctate localization is observed in only 26% of expressing cells, whereas most cells show diffuse expression throughout the cytoplasm with occasional weak concentration in the inner segment (brackets in Fig. 7B and B′, quantification in Fig. 7C), indicating that Cc2d2a is at least partially required for punctate localization of Rab8 in the inner segment. To test whether Cc2d2a localization is dependent on Rab8, we used a previously described splice-blocking rab8 morpholino (71) which causes a sinusoidal body shape and a retinal phenotype (shortened outer segments and rhodopsin mislocalization) similar to cc2d2a mutants (Supplementary Material, Fig. S3A–D). Despite this retinal phenotype in Rab8 morphants, Cc2d2a localization at the base of photoreceptor cilia is unaffected (Fig. 7D). Together, these data indicate that Rab8 localization depends on Cc2d2a, whereas Cc2d2a localization occurs independently of Rab8.

cc2d2a is required for Rab8 localization and interacts genetically with rab8 in ciliated cells. (A–B′) Expression of a transducin-promoter-driven Rab8-cherry construct in cc2d2a+/+ photoreceptors (A and A′) is concentrated in one punctum at the base of the outer segment (arrows), whereas it is diffuse in cc2d2a−/− photoreceptors (B and B′, bracket). (C) Proportion of Rab8-cherry expressing photoreceptors with punctate versus slightly enhanced inner segment or diffuse cellular expression (***P < 0.0001, χ2 test, bars indicate 95% confidence interval). (D) Cc2d2a localization (red) in 4 d.p.f. Rab8 morphant (rab8MO) retinas co-stained with acetylated tubulin to mark cilia. (E–J) Synergistic effect of partial rab8 knockdown on the retinal phenotype of cc2d2a−/− embryos. (E–G) Length of outer segments highlighted with bodipy counterstain (red, brackets) is reduced in rab8-morpholino-injected cc2d2a mutant embryos (G) compared with control-morpholino-injected mutants (F). At the same dose of rab8 morpholino, cc2d2a+/+ embryos have well-formed outer segments of normal length (E). (H–J) Rhodopsin localization (4D2 antibody, red) is virtually absent from remnant outer segments in rab8-morpholino-injected cc2d2a mutants (J) compared with control-morpholino-injected mutants (I). At the same dose of rab8 morpholino, there is no mislocalization of rhodopsin in cc2d2a+/+ siblings (H). (K) Histogram showing the proportion of embryos with pronephric cysts (red bars indicate cc2d2a−/− mutants and blue bars indicate wild-type and heterozygote siblings). Bars indicate 95% confidence intervals. Pairwise comparisons between rab8-morpholino-injected cc2d2a−/− fish and uninjected siblings, as well as rab8-morpholino-injected cc2d2a−/− and control (gbx2) morpholino-injected cc2d2a−/− fish are significant (**P = 0.0001, χ2 test). NS, non-significant. All images are cryosections of 5 d.p.f. retinas except (D), which is a cryosection of a 4 d.p.f. retina. Scale bars are 4 µm in all panels. +/+ or +/− in (E) and (H) refers to cc2d2a+/+ or +/−.
cc2d2a interacts genetically with rab8 in ciliated cells
Given the effect of Cc2d2a loss on Rab8 localization, we investigated a potential functional relationship in vivo between cc2d2a and rab8, using a morpholino knockdown approach in our cc2d2a mutant background. Using low doses of rab8 morpholino that do not cause a phenotype in wild-type (cc2d2a+/+ or +/−) siblings, we observe a significant enhancement of the retinal phenotype in 4d.p.f. cc2d2a−/− fish based on outer segment length and rhodopsin mislocalization. To rule out non-specific enhancement of the retinal phenotype, we injected a control morpholino that effectively targets a gene involved in an unrelated developmental process (gbx2). Average outer segment length measured on bodipy-stained sections is significantly shorter (P < 0.0001, Student's t-test) in rab8-morpholino-injected cc2d2a−/− embryos (n = 118 photoreceptors from 10 embryos) compared with uninjected or control-morpholino-injected cc2d2a−/− embryos (n = 108 photoreceptors from 9 embryos) (Fig. 7E–G and Supplementary Material, Fig. S3F). Moreover, rab8-morpholino-injected mutants can be distinguished from control-injected mutants based on rhodopsin mislocalization, since partial knock-down of rab8 in cc2d2a mutants abolishes localization of rhodopsin to remnant outer segments (Fig. 7H–J).
To test whether this functional interaction between cc2d2a and rab8 is relevant in other ciliated cell types, we took advantage of the incomplete penetrance of the pronephric cyst phenotype in cc2d2a mutants, whereby 30–50% of cc2d2a−/− fish develop pronephric cysts. Using sub-phenotypic doses of rab8 morpholino, we observe an increase in the proportion of cc2d2a mutants that develop pronephric cysts (53% in uninjected and 50% in control-morpholino-injected embryos versus 89% in rab8-morpholino-injected embryos, P = 0.0001, χ2 test, Fig. 7K), thereby indicating that this functional interaction occurs in multiple cell types.
DISCUSSION
CC2D2A mutations cause Joubert syndrome in ~10% of patients, a subset of whom have retinal dystrophy (30,51). Our work using a zebrafish mutant is the first model for CC2D2A-related retinal dystrophy, confirming a role for CC2D2A in the development and function of the photoreceptor outer segment and providing an opportunity to explore the mechanisms underlying the retinal dystrophy seen in patients with Joubert syndrome. Our findings indicate a role for Cc2d2a in vesicle trafficking of transmembrane outer segment components such as opsins, based on the striking accumulation of vesicles and mislocalization of opsins in mutant photoreceptors. In addition, we show that loss of Cc2d2a leads to partial mislocalization of Rab8, a key regulator of trafficking and fusion of opsin-carrier vesicles at the periciliary ridge complex involved in outer segment disk morphogenesis and ciliary membrane biogenesis (21–23). Further supporting a role for Cc2d2a in vesicle trafficking, we demonstrate a functional interaction with rab8 in photoreceptors and other ciliated cell types. Based on these findings, we propose a model where Cc2d2a protein, localized at the transition zone/connecting cilium of photoreceptors, facilitates docking and fusion of opsin carrier vesicles via Rab8 localization, thereby acting as an ‘entry-facilitator’ for cilia-directed proteins. An alternate model, in which defective ciliogenesis underlies the observed abnormal outer segment development, is unlikely, based on the presence of normal-appearing cilia in multiple mutant tissues where Cc2d2a is normally expressed, including the kidney and retina, despite phenotypic anomalies in these organs. This indicates that Cc2d2a is not essential for ciliogenesis and that loss of cilia per se is not the primary mechanism underlying the photoreceptor phenotype observed in cc2d2a mutants. Moreover, zebrafish ift88 and ift172 mutants that lack cilia entirely do not display the same massive vesicle accumulation in photoreceptors as cc2d2a mutants (9). Therefore, vesicle accumulation is not simply a consequence of deficient outer segment formation, but is specific to loss of Cc2d2a function, thereby supporting a role for Cc2d2a in vesicle trafficking and fusion.
A role in Rab8-dependent vesicle trafficking has been shown for other ciliopathy-related proteins or protein complexes including CEP290, AHI1, RPGR or the BBSome (23,42,43,72). Indeed, CEP290, AHI1 and RPGR si/shRNA-treated cells lose ciliary localization of Rab8, and AHI1 shRNA-treated cells display abnormal vesicle trafficking (42). In addition, the photoreceptor phenotype in CEP290- and AHI1-deficient mice is similar to what we observe in cc2d2a zebrafish mutants: abnormal outer segments, but normal connecting cilia, and opsin trafficking defects (45,47). These phenotypic similarities suggest a shared role in opsin transport further supported by the comparable effects on Rab8 localization. Cc2d2a could provide a docking point for Rab8 at the base of the cilium, thereby facilitating subsequent Rab8-mediated fusion of vesicles with the periciliary membrane. This model is consistent both with Cc2d2a localization at the base of the cilium and with the punctate Rab8 localization, which is lost in cc2d2a mutant photoreceptors. This docking function could be achieved either through a direct physical interaction with Rab8 or, more likely, through participation in a complex of ciliopathy proteins required for Rab8 localization. In fact, previous evidence supports the idea that Cc2d2a likely functions together with Cep290 to localize Rab8 in photoreceptors, since CEP290 is required for Rab8 localization in cultured cells (43), co-localizes with CC2D2A at the base of cilia and interacts physically and genetically with Cc2d2a (30).
The relationship between Cc2d2a, Rab8 and vesicle trafficking is undoubtedly more nuanced than a linear Cc2d2a-dependent pathway for Rab8 localization would suggest, given that Rab8 can localize normally in a minority of cc2d2a mutant photoreceptors, and that Rab8 knock-down enhances the photoreceptor and kidney phenotypes of cc2d2a−/− embryos. Rab8 distribution in ciliated cells is complex, having been detected in the Golgi, the basal body and the cilium in different cell types under different conditions (21,23,42,43,73), and likely depends on multiple factors in addition to Cc2d2a; furthermore, while Rab8 is essential for cilium formation (23,74), we do not observe fundamental defects in ciliogenesis in cc2d2a mutants. Thus, Cc2d2a likely plays a selective role, facilitating only certain aspects of Rab8 localization and function.
While our data strongly support an indirect role for Cc2d2a in trafficking through Rab8, they do not rule out a separate direct role for Cc2d2a in vesicle docking and fusion, or other additional functions. A recent work in C. elegans indicates that CC2D2A localizes at the transition zone of amphid and phasmid cilia and shows a role for CC2D2A, together with other ciliopathy proteins, as a gate-keeper of the cilium, based on ectopic localization of transmembrane proteins in the cilia of mutant worms (41). A similar localization and function has been described for CEP290 in Chlamydomonas (39), leading to a model in which transition zone proteins restrict the entry of non-ciliary proteins into the cilium. This gate-keeper function differs from the entry-facilitator function for Cc2d2a identified by our work, in that the former is required to keep non-ciliary-directed proteins out of the cilium, whereas the latter is actively required for the entry of ciliary proteins. Loss of an entry-facilitator thus results in lack of normal transport of components that should normally enter the compartment and mislocalization of these components to the cell (i.e. rhodopsin in cc2d2a mutant photoreceptors). In contrast, loss of a gate-keeper results in the entry of non ciliary-components into the ciliary compartment. While we did not test directly whether Cc2d2a restricts the access of proteins to the outer segment in zebrafish photoreceptors, our model whereby Cc2d2a is required for docking/fusion of specific vesicles to the base of the cilium does not rule out a potential additional role as a gate-keeper. In fact, proteins localized at the entry check-point of cilia may play a double role: assisting cilium-targeted vesicles to dock/fuse and also preventing non-cilium-targeted components from entering the cilium. One example for such a dual function is provided by CEP290, which has been shown to act as a gate-keeper in Chlamydomonas, and which had previously been shown to control Rab8 localization to the cilium, and therefore to be at least indirectly involved in vesicle trafficking (39,43).
The emerging model thus identifies the basal body/transition zone as a hub controlling access to the cilium, where ciliopathy proteins localized at this entry-point facilitate the selective entry of ciliary-bound proteins through a role in vesicle trafficking (entry-facilitator function) (23,42,45) and restrict access to the cilium to non-ciliary-targeted proteins (gate-keeper function) (39,41). Many of the proteins residing at this hub function together in complexes or modules to direct the incoming cargo (38,41). Our findings place Cc2d2a at this hub in a vertebrate model with direct relevance to a human ciliopathy disease state and support an active role for Cc2d2a as an entry-facilitator, in addition to the recently described function as a gate-keeper. Future work will focus on how Cc2d2a and other transition zone proteins coordinate with the BBSome and the intraflagellar transport particle to successfully transport transmembrane proteins to the cilium.
MATERIALS AND METHODS
Animals
Zebrafish (Danio rerio) were maintained as described (75). The cc2d2aw38/sentinel mutant (referred to simply as ‘cc2d2a mutant’ or cc2d2a−/− throughout) was originally described in the work of Owens et al. (50). The ift88/polaris mutant was originally described in the work of Doerre and Malicki (54) and was a gift from Brian Perkins (Texas A&M). cc2d2aw38 mutant embryos were obtained from cc2d2a heterozygote incrosses and genotyped using dcap primers (forward: 5′-GAGAGACTGCAGGACGAGCAGGA; reverse: 5′-TAGCCGTACCTGTTTTCTTTTCCAGGATAT). A minority (~10%) of homozygous cc2d2a mutant larvae developed swim-bladders and were raised to adulthood. Maternal zygotic cc2d2a embryos were generated through in vitro fertilization using eggs from homozygous cc2d2a mutant females fertilized with sperm from cc2d2a heterozygous males. Embryos were raised at 28.5°C. In some cases, pigment development was inhibited by the use of phenylthiourea as described in Westerfield (75). All of the animal protocols for the use of zebrafish in the research described in this paper are in compliance with internationally recognized guidelines for the use of fish in biomedical research. The protocols were approved by the Fred Hutchinson Cancer Research Center Institutional Animal Care and Use Committee (file no. 1392).
Electroretinograms
ERGs were done as described previously (58). Briefly, 5 and 6 d.p.f. old larvae were anesthetized in tricaine and eyes were removed using a fine tungsten wire loop. Excised eyes were then placed in an oxygenated Ringer's solution (in mm; 130 NaCl, 2.5 KCl, 20 NaHCO3, 0.7 CaCl2, 1.0 MgCl2 and 20 glucose), and a glass electrode was positioned directly onto the cornea. After 5 min of dark adaptation, eyes were exposed to white light flashes of increasing intensities for 1 ms and their electrical response was recorded. Data were acquired and processed as described previously (76).
Zebrafish histology and electron microscopy
Plastic sections were generated as described previously (77). Fixation was in 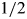 strength Karnovsky's [2% paraformaldehyde (PFA), 2.5% gluteraldehyde] overnight at 4°C. Blocks were sectioned at 1 µm thickness up to the optic nerve for plastic sections and the same blocks were further sectioned at 70 nm thickness and stained with uranyl acetate and lead citrate for transmission electron microscopy, which was performed using a JEOL 1230 microscope. Cryosections were performed as follows: 5d.p.f. larvae were fixed in 4% PFA [5 h at room temperature (RT) or overnight at 4°C], or with 2% trichloroacetic acid (TCA) (3 h at RT), subsequently embedded in OCT (Tissue-Tek) as previously described (75) and cryosectioned at 10 µm thickness.
strength Karnovsky's [2% paraformaldehyde (PFA), 2.5% gluteraldehyde] overnight at 4°C. Blocks were sectioned at 1 µm thickness up to the optic nerve for plastic sections and the same blocks were further sectioned at 70 nm thickness and stained with uranyl acetate and lead citrate for transmission electron microscopy, which was performed using a JEOL 1230 microscope. Cryosections were performed as follows: 5d.p.f. larvae were fixed in 4% PFA [5 h at room temperature (RT) or overnight at 4°C], or with 2% trichloroacetic acid (TCA) (3 h at RT), subsequently embedded in OCT (Tissue-Tek) as previously described (75) and cryosectioned at 10 µm thickness.
Antibody generation
The mouse monoclonal antibody against Cc2d2a was generated in the FHCRC (Fred Hutchinson Cancer Research Center) Hybridoma Production Facility by immunizing mice with a GST fusion to zebrafish Cc2d2a protein (amino acids 430–1019). Hybridomas were generated from spleens of these mice to obtain the monoclonal P2D9 line (IgG1 subtype). Supernatant from the extinction culture was collected and used for western blotting and immunofluorescence.
Cloning, cell culture and transfections, western blots
cc2d2a was amplified from wild-type cDNA and cloned in the pCSegfpDest vector (78) using the Gateway System (Invitrogen). HEK293T cells were grown in DMEM (Sigma) supplemented with 10% FBS (Hyclone). GFP-tagged wild-type and W628* Cc2d2a were expressed in HEK293T cells following transfection with Lipofectamine2000 (Invitrogen). Western blots were performed on lysates from these transfected HEK293T cells obtained 24 h after transfection as previously described (79).
Western blots were performed on lysates from wild-type, zygotic and mz 2d.p.f. embryos as follows: embryos were deyolked by trituration in 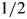 Ginzburg followed by homogenization in NP-40 lysis buffer supplemented with PMSF and a protease inhibitor cocktail (Roche). Ten micrograms of total protein per lane was run on a 4–12% NuPAGE gel under denaturing conditions followed by transfer onto PVDF membrane (Invitrogen). Immunoblotting was performed using the mouse anti-Cc2d2a P2D9 (1:20) and mouse anti-alpha-tubulin (1:2000, Sigma T6199) antibodies.
Ginzburg followed by homogenization in NP-40 lysis buffer supplemented with PMSF and a protease inhibitor cocktail (Roche). Ten micrograms of total protein per lane was run on a 4–12% NuPAGE gel under denaturing conditions followed by transfer onto PVDF membrane (Invitrogen). Immunoblotting was performed using the mouse anti-Cc2d2a P2D9 (1:20) and mouse anti-alpha-tubulin (1:2000, Sigma T6199) antibodies.
Immunohistochemistry and whole-mount antibody staining
Whole-mount antibody staining was performed on zebrafish embryos fixed with 4% PFA (5 h at RT or overnight at 4°C), MeOH (80% MeOH/20% DMSO; overnight at 4°C) or with 2% TCA (3 h at RT). Embryos were permeabilized with acetone at −20°C for 7 min (only for PFA-fixed embryos), washed and blocked using PBDT (PBS, 1% DMSO, 0.1% Triton, 2 mg/ml BSA) with 5% goat serum for 30 min at RT before incubation with primary antibody in PBDT with 2% goat serum overnight at 4°C. Secondary antibodies were fluorochrome-conjugated Alexa Fluor 488 or 594 goat anti-mouse or goat anti-rabbit IgG (Molecular Probes) used at 1:300. Primary antibodies were used at the following dilutions: mouse cc2d2a 1:20, rabbit arl13b 1:200 (gift from Z. Sun, Yale University) (11), mouse gamma-tubulin 1:1000 (Sigma), mouse acetylated tubulin 1:1000 (Sigma). Immunohistochemistry on zebrafish retinal cryosections was performed with a similar protocol as for whole mounts except that no acetone permeabilization was performed. Antibody dilutions for immunohistochemistry were: mouse cc2d2a 1:20, rabbit arl13b 1:200 (gift from Z. Sun, Yale University) (11), rabbit gamma-tubulin 1:500 (Sigma), mouse acetylated tubulin 1:500 (Sigma), 4D2 1:200 (gift from R. Molday, University of British Colombia) (80), 4c12 1:100 (gift from James Fadool, Florida State University) (55), zpr1 1:100 (ZIRC) (52), BOPS 1:160 (gift from D. Hyde, University of Notre Dame) (81), rabbit transducin (S. Brockerhoff) (66), ift88 1:5000 (gift from B. Perkins, Texas A&M) (8). Prolong Gold Anti-fade (Molecular Probes) was used for mounting the sections and whole mounts. Confocal imaging was performed on an LSM5 Pascal microscope (Carl Zeiss) or on an Axio Observer Z1 microscope (Carl Zeiss) equipped with a Yokogawa spinning disk with ×40 and ×63 objectives.
Morpholino injections
Morpholino oligonucleotides (MO) from Genetools, Inc., were resuspended in distilled RNAse free water and diluted to the appropriate concentration in phenol red 0.25% to allow optimal visualization and consistency of bolus size. At the 1–2 cell stage, 1 nl of morpholino at the appropriate concentration was injected into zebrafish embryos. The Rab8 morpholino was previously described (82). As a control, we used morpholinos against the homeobox gene gbx2 (83), at doses that cause significant toxicity in the form of CNS cell death (2 ng). Scoring for pronephric cysts, outer segment length and rhodopsin mislocalization was performed blinded as to the injection status of the larvae.
Transgenic constructs and fish lines
For the generation of the Rab8-cherry fusion construct, Danio rerio rab8a (NM_001089562) was isolated by RT-PCR, sequence-confirmed and used to create an in-frame N-terminal fusion with mCherry driven by the cone-specific transducin promoter (53). All recombineering was done using entry plasmids from the Tol2kit clone (84). The gnat2:Man2a-RFP construct, the tg(TaCP:MCFP) zebrafish line and the tg(arl13b-GFP) zebrafish line were originally described in the work of Insinna et al. (70), Lewis et al. (53) and Borovina et al. (85), respectively.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institute of Health grants (HD37909 to C.B.M., 5KL2RR025015 and R01NS064077 to D.D., R01EY014167 to B.A.L., R01EY018814 to G.S. and S.E.B. and P30EY01730 to G.S.); and a Basil O'Connor Starter Scholar Award from the March of Dimes to D.D. C.B.M. is an investigator with the Howard Hughes Medical Institute. R.B.-G. was supported by an National Institute of Health Ruth L. Kirschstein NRSA Medical Genetics Postdoctoral Fellowship (5T32GM007454). Funding to pay the Open Access publication charges for this article was provided by Howard Hughes Medical Institute (HHMI).
ACKNOWLEDGEMENTS
We wish to thank Zhaoxia Sun, Brian Perkins, David Hyde, Robert Molday, Brian Ciruna and James Fadool for kindly providing antibodies and fish lines. We thank members of the Moens Laboratory and Ronald Roepman for helpful discussions during the course of this work. We also thank Edward Parker for assistance with light microscopy of plastic sections, Bobbie Schneider and Steve MacFarlane (FHCRC shared resources) for assistance with electron microscopy, Elizabeth Wayner and the FHCRC antibody development facility for antibody development and Sean Rhodes for expert fish care.
Conflict of Interest statement. None declared.
REFERENCES
Articles from Human Molecular Genetics are provided here courtesy of Oxford University Press
Full text links
Read article at publisher's site: https://doi.org/10.1093/hmg/ddr332
Read article for free, from open access legal sources, via Unpaywall:
https://academic.oup.com/hmg/article-pdf/20/20/4041/17253515/ddr332.pdf

Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Discover the attention surrounding your research
https://www.altmetric.com/details/102355161
Smart citations by scite.ai
Explore citation contexts and check if this article has been
supported or disputed.
https://scite.ai/reports/10.1093/hmg/ddr332
Article citations
Astrogliosis and neuroinflammation underlie scoliosis upon cilia dysfunction.
Elife, 13:RP96831, 10 Oct 2024
Cited by: 1 article | PMID: 39388365 | PMCID: PMC11466456
Male germ cell-associated kinase is required for axoneme formation during ciliogenesis in zebrafish photoreceptors.
Dis Model Mech, 17(7):dmm050618, 16 Jul 2024
Cited by: 0 articles | PMID: 38813692 | PMCID: PMC11273301
Association of genetic variation in COL11A1 with adolescent idiopathic scoliosis.
Elife, 12:RP89762, 26 Jan 2024
Cited by: 0 articles | PMID: 38277211 | PMCID: PMC10945706
The role of cilia during organogenesis in zebrafish.
Open Biol, 13(12):230228, 13 Dec 2023
Cited by: 0 articles | PMID: 38086423 | PMCID: PMC10715920
Review
 Free full text in Europe PMC
Free full text in Europe PMCRetinal degeneration in rpgra mutant zebrafish.
Front Cell Dev Biol, 11:1169941, 07 Jun 2023
Cited by: 0 articles | PMID: 37351277 | PMCID: PMC10282147
Go to all (76) article citations
Data
Data behind the article
This data has been text mined from the article, or deposited into data resources.
BioStudies: supplemental material and supporting data
Diseases (3)
- (2 citations) OMIM - 213300
- (1 citation) OMIM - 266900
- (1 citation) OMIM - 209900
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
Loss-of-function of the ciliopathy protein Cc2d2a disorganizes the vesicle fusion machinery at the periciliary membrane and indirectly affects Rab8-trafficking in zebrafish photoreceptors.
PLoS Genet, 13(12):e1007150, 27 Dec 2017
Cited by: 16 articles | PMID: 29281629 | PMCID: PMC5760100
The Ciliopathy Protein CC2D2A Associates with NINL and Functions in RAB8-MICAL3-Regulated Vesicle Trafficking.
PLoS Genet, 11(10):e1005575, 20 Oct 2015
Cited by: 53 articles | PMID: 26485645 | PMCID: PMC4617701
The Ciliopathy Gene ahi1 Is Required for Zebrafish Cone Photoreceptor Outer Segment Morphogenesis and Survival.
Invest Ophthalmol Vis Sci, 58(1):448-460, 01 Jan 2017
Cited by: 27 articles | PMID: 28118669 | PMCID: PMC5270624
The photoreceptor cilium and its diseases.
Curr Opin Genet Dev, 56:22-33, 28 Jun 2019
Cited by: 28 articles | PMID: 31260874
Review
Funding
Funders who supported this work.
Howard Hughes Medical Institute
NCRR NIH HHS (1)
Grant ID: 5KL2RR025015
NEI NIH HHS (6)
Grant ID: R01EY018814
Grant ID: P30 EY001730
Grant ID: R01EY014167
Grant ID: R01 EY018814-03
Grant ID: P30EY01730
Grant ID: R01 EY018814
NICHD NIH HHS (1)
Grant ID: HD37909
NIGMS NIH HHS (1)
Grant ID: 5T32GM007454
NINDS NIH HHS (2)
Grant ID: R01NS064077
Grant ID: R01 NS064077


