Abstract
Free full text
Dynactin helps target Polo-like kinase 1 to kinetochores via its left-handed beta-helical p27 subunit
Abstract
Dynactin is a protein complex required for the in vivo function of cytoplasmic dynein, a microtubule (MT)-based motor. Dynactin binds both dynein and MTs via its p150Glued subunit, but little is known about the ‘pointed-end complex’ that includes the protein subunits Arp11, p62 and the p27/p25 heterodimer. Here, we show that the p27/p25 heterodimer undergoes mitotic phosphorylation by cyclin-dependent kinase 1 (Cdk1) at a single site, p27 Thr186, to generate an anchoring site for polo-like kinase 1 (Plk1) at kinetochores. Removal of p27/p25 from dynactin results in reduced levels of Plk1 and its phosphorylated substrates at kinetochores in prometaphase, which correlates with aberrant kinetochore–MT interactions, improper chromosome alignment and abbreviated mitosis. To investigate the structural implications of p27 phosphorylation, we determined the structure of human p27. This revealed an unusual left-handed β-helix domain, with the phosphorylation site located within a disordered, C-terminal segment. We conclude that dynactin plays a previously undescribed regulatory role in the spindle assembly checkpoint by recruiting Plk1 to kinetochores and facilitating phosphorylation of important downstream targets.
Introduction
During interphase, dynein, a microtubule (MT)-associated, minus end-directed motor, and dynactin, its ≈1 MDa partner complex, function primarily to move membrane-bound organelles and other intracellular components. During mitosis, the dynein/dynactin assembly plays a completely different set of roles. It is essential for spindle pole focusing, helps chromosomes engage with and move on spindle MTs, and is believed to facilitate spindle checkpoint silencing by transporting regulators away from kinetochores (Echeverri et al, 1996; Howell et al, 2000; Yang et al, 2007; Bader and Vaughan, 2010).
MDa partner complex, function primarily to move membrane-bound organelles and other intracellular components. During mitosis, the dynein/dynactin assembly plays a completely different set of roles. It is essential for spindle pole focusing, helps chromosomes engage with and move on spindle MTs, and is believed to facilitate spindle checkpoint silencing by transporting regulators away from kinetochores (Echeverri et al, 1996; Howell et al, 2000; Yang et al, 2007; Bader and Vaughan, 2010).
Dynactin comprises 11 different polypeptide components, each present in one or more copies, which are organized into three structural/functional domains. The structural core of dynactin is a short copolymer of the actin family proteins Arp1, Arp11 and actin, plus the actin capping protein, CP (CapZ). This central structure is flanked by distinct structures at each end. The shoulder/sidearm assembly (p150Glued, dynamitin and p24 subunits), which envelops the ‘barbed’ end of the Arp polymer (Imai et al, manuscript in preparation), binds to dynein and MTs. Located at the opposite, ‘pointed’ end is the p62 subunit and the p27/ p25 heterodimer (Schafer et al, 1994; Eckley et al, 1999). The Arp polymer and the pointed end complex (p27/p25 plus p62 and Arp11) are assumed to participate in cargo binding (Schroer and Cheong, 2012) and specification (Yeh et al, 2012) although the molecular details remain undefined.
Of all dynactin subunits, those that comprise the pointed end complex are most mysterious. The p62, p27 and p25 components appear to be dispensable for minimal dynactin function as they are absent in yeast. The p62 subunit, which is required for dynactin stability, contains a predicted Zn2+ binding, RING/LIM-like motif near the N-terminus and, along with its closely associated subunit Arp11, contributes to dynactin targeting to the nuclear envelope (Yeh et al, 2012). The two smallest dynactin subunits, p27 and p25, are important dynactin and dynein binding to endomembranes (Zhang et al, 2011; Yeh et al, 2012). A mitotic role for the p27/p25 heterodimer has not been explored.
In this study, we investigated the function of dynactin’s p27 component in mitosis. Using RNA interference (RNAi), we determined that p27 and p25, which are co-depleted by small-interfering RNA (siRNAs) to either, are required for normal mitotic timing. Cells depleted of p27 and p25 show defects in kinetochore/MT interactions and chromosome alignment, but unlike cells lacking other dynactin subunits that also exhibit these behaviours, they show accelerated progression through mitosis. The mitotic function of p27 depends upon cyclin-dependent kinase 1 (Cdk1) phosphorylation at Thr186, which generates a binding site for the important mitotic kinase, polo kinase 1 (Plk1). Depletion of p27 and p25 does not interfere with formation of mitotic spindles, but Plk1 levels are significantly reduced at kinetochores. In conjunction with our functional work, we solved the crystal structure of human recombinant p27 at 2.2Å resolution. The molecular model revealed a unique left-handed β-helix domain, the first such tertiary fold to be characterized in a vertebrate protein, and showed that the Cdk1 phosphorylation site is in the unstructured, C-terminal segment. Our work reveals that dynactin serves as a binding platform for dynein and Plk1 at kinetochores that governs passage through the spindle assembly checkpoint.
Results
Dynactin p27 and p25 are required for normal mitotic timing
To investigate whether p27 and p25 might play roles in mitosis, we removed them from dynactin using RNAi. Treatment of Cos7 fibroblast cells with p27 siRNAs results in depletion of both p27 and p25, but leaves the rest of the dynactin complex intact (Yeh et al, 2012). To compare the effects of p27/p25 depletion with general impairment of dynactin function, we performed parallel experiments in which p150Glued, the dynein-binding component of dynactin, was depleted via RNAi. As a final control, we evaluated the effect of depleting Arp11, which leads to the loss of the entire dynactin molecule (Yeh et al, 2012). As expected, depletion of either p150Glued or Arp11 resulted in profound spindle defects (Yeh et al, 2012; Figure 6C, MTs), pseudoprometaphase arrest and an increase in mitotic index (Figure 1A and B). By contrast, depletion of p27 and p25 yielded cells whose spindle structure appeared normal (Yeh et al, 2012; Figure 6C). We were surprised to find that the mitotic index of the p27/p25-depleted population was reduced (3.1 versus 5.6% for controls; Figure 1B) due to a reduction in the proportion of Cos7 cells at all stages of mitosis (Figure 1A). Closer inspection of mitotic cells revealed lagging chromosomes and misaligned metaphase plates, suggesting chromosome congression defects (Figure 1C and D). Expression of siRNA-resistant, mouse p27, which rescues both p27 and p25 (Yeh et al, 2012) and yields their full incorporation into dynactin (Supplementary Figure 1A), restored the mitotic index to normal (Figure 1A and B), indicating that the defects were not due to off-target effects of the p27 siRNAs.

Effects of dynactin subunit depletion on mitotic progression. (A through C) Analysis of mitotic figures in asynchronous Cos7 populations stained for phospho-histone H3 (A, B) or DAPI (C). (A) Mitotic progression in cells transfected with dynactin subunit siRNAs alone or in conjunction with p27 rescue plasmids. Mitotic stages were determined on the basis of chromosome configuration (mean±s.d., three experiments, n>6000 cells). Mitotic cells exhibiting incomplete chromosome congression were scored as being in prometaphase. (B) Mitotic indices of cell populations transfected with dynactin subunit siRNAs (as in A; mean±s.d., three experiments, n>6000 cells). (C) Representative images of chromosome alignment phenotypes in metaphase (left) and anaphase (right) cells in the control and p27/p25-depleted population. Bar=5μm. (D) Mitotic populations of cells treated with dynactin subunit siRNAs were scored for the presence of misaligned or lagging chromosomes (as in C; mean±s.d., three experiments, n>600 mitotic cells). (E) Images of living control and p27/p25-depleted cells expressing histone 2B-mCherry starting 6min prior to nuclear envelope breakdown (0:00 time stamp) and continuing through anaphase. Arrows indicate examples of uncongressed or lagging chromosomes. Twenty-five cells were filmed in each condition, and 76% (19/25 cells) of p27/p25-depleted cells had uncongressed or lagging chromosomes. Bar=5μm. (F) The elapsed time (mean±s.d.; n=25 cells per condition) determined from live-cell recordings (as in E and Supplementary Movies) between nuclear envelope breakdown and anaphase onset in control and p27/p25-depleted cells with and without p27 rescue plasmids.
Source data for this figure is available on the online supplementary information page.
The combination of chromosome alignment defects and a reduced number of cells in mitosis suggests that cells are proceeding through mitosis more rapidly than normal, due to lack of activation of the spindle assembly checkpoint in the face of defects (i.e., checkpoint bypass) (Fava et al, 2011). This behaviour is markedly different from the pseudoprometaphase arrest and prolonged checkpoint activation that is typical of dynein or dynactin perturbation (Echeverri et al, 1996; Howell et al, 2000). Live imaging, either via phase contrast (Supplementary Movies 7 and 8) or of cells expressing mCherry-tagged histone 2B, revealed that cells depleted of p27 and p25 spend significantly less time in the interval between nuclear envelope breakdown (NEBD) and anaphase (28.1±2.2min versus 41.7±2.4min for controls; Figure 1E and and1F;1F; Supplementary Movies 1 and 2). Instead of pausing in metaphase for a few minutes, the cells initiated anaphase as soon as the bulk of chromosomes had converged into a single mass even though uncongressed chromosomes were still observed (Figure 1E). By contrast, cells expressing siRNA-resistant mouse p27 remained in metaphase for a few minutes and exhibited a normal interval between NEBD and anaphase (42±2.3min, Figure 1F; Supplementary Movies 3 and 9). The lack of a ‘wait-anaphase’ period and the presence of uncongressed chromosomes strongly suggest that cells depleted of p27 and p25 do not execute the spindle assembly checkpoint effectively. Treatment of p27/p25-depleted cells with nocodazole was found to result in mitotic arrest (mitotic index 64.5±4.2 versus 65.4±4.2% for control; Supplementary Figure 2), indicating that the checkpoint was operational, however, it was not activated in cells depleted of p27 and p25. Chromosome alignment in cells depleted of Arp11 or p150Glued was aberrant and mitosis was highly prolonged (Supplementary Movies 5 and 6), in agreement with our analysis of fixed cells (Figure 1A–D).
Efficient and accurate chromosome congression depends upon the presence of both dynein and dynactin at kinetochores (Starr et al, 1998; Yang et al, 2007). Recruitment involves an interaction of dynactin’s dynamitin component with the RZZ (Rod/ZW10/Zwilch) complex (Starr et al, 1998) plus a number of other interactions. p27/p25 is required for dynactin association with endosomes and dynein-based endosome movement (Zhang et al, 2011; Yeh et al, 2012), but whether these two dynactin components also contribute to kinetochore binding and dynein recruitment to kinetochores is unknown. To explore these questions, we quantified the levels of dynactin’s p150Glued, Arp1 and p27 components, plus dynein (dynein intermediate chain, DIC) in cells treated with dynactin subunit siRNAs (Figure 2). Cells depleted of either p150Glued or the entire dynactin molecule (via Arp11 depletion) showed a significant reduction in kinetochore-associated dynactin. Depletion of p27 and p25, by contrast, left kinetochore dynactin in place. Free, exogenous p27 expressed at low levels (via the siRNA-resistant p27 rescue vector) in cells depleted of p150Glued or Arp11 did not target to kinetochores (Supplementary Figure 3), indicating that p27 does not bind this site unless it is incorporated into dynactin. Depletion of p150Glued or Arp11 also caused significant reduction in dynein at kinetochores, whereas p27 depletion did not (Figure 2A and B). This result agrees with previous reports that dynein binding to kinetochores involves dynactin’s p150Glued component (Echeverri et al, 1996; Whyte et al, 2008). Levels of the kinetochore components ZW10, Bub1, BubR1, Bub3, MPS1 and the dynein/dynactin interactor, Spindly (Liu et al, 2006; Griffis et al, 2007; Liang et al, 2007; Whyte et al, 2008; Chan et al, 2009; Gassmann et al, 2010) were unchanged under all three depletion conditions (Figure 2C; Supplementary Figure 4) confirming that none of these proteins depend upon dynein or dynactin for kinetochore binding and verifying that kinetochore structure was not grossly perturbed. This analysis demonstrates that kinetochores in cells depleted of p27 and p25 still contain normal steady-state levels of both dynein and dynactin, with the only notable difference being the absence of p27 and p25 from dynactin. This system thus permits study of the function of p27 and p25 at kinetochores selectively, without concerns about possible non-specific or downstream consequences of loss of dynactin or dynein.

Localization of dynein, dynactin and other proteins to prometaphase kinetochores. (A) Representative images from asynchronous Cos7 populations were fixed and stained for Arp1, p150Glued, p27 or dynein intermediate chain (DIC). Bar=5μm. (B) Fluorescence intensities at prometaphase kinetochores of cells as in (A) were quantified, normalized to anti-centromere antigen (ACA) pixel values and expressed as per cent of controls (mean±s.e.m., n>600 kinetochores in 15 cells). (C) The fluorescence intensities at prometaphase kinetochores of the proteins indicated were quantified and normalized as in (B) (see Supplementary Figure 4 for images).
Source data for this figure is available on the online supplementary information page.
Depletion of p27 and p25 from kinetochore dynactin appears to interfere with normal execution of the spindle assembly checkpoint, a phenomenon of great interest in which many molecules have been implicated. Among these are the mitotic checkpoint complex (MCC) and the mitotic regulatory kinase, Plk1. Both dynactin and dynein are reported substrates of Plk1 and two dynactin components, p150Glued and dynamitin, are components of the polo-binding interactome, suggesting an interaction with Plk1 (Lowery et al, 2007; Li et al, 2010; Bader et al, 2011). Whether the dynactin/Plk1 interaction is direct, and if so, which dynactin component is involved, has not been explored.
Dynactin p27 is phosphorylated by Cdk1 and binds Plk1
Plk1 binding commonly requires priming phosphorylation by Cdk1. In search of regulatory mechanisms that might involve p27 or p25 phosphorylation, we searched their amino-acid sequences for predicted phosphorylation motifs. The human p27 sequence contains a single consensus Cdk1 phosphorylation site, Thr186, that is conserved among vertebrates (Figure 3A). Phosphorylation at this site creates a consensus Plk1 binding site (Elia et al, 2003). Although p25 is somewhat related to p27, no p25 species we examined contained a Cdk1 consensus sequence or a predicted polo-binding motif. To verify that p27 could serve as a substrate for Cdk1, we performed in vitro phosphorylation using purified bovine and embryonic chick brain dynactin (Figure 3B). p27 was found to be a major target, along with p150Glued. We then used point mutagenesis to verify that p27 Thr186 was the Cdk1 phosphorylation site. For this work, we utilized purified, recombinant p27/p25 heterodimers to mimic p27 in its native context. Wild-type p27, along with two variants, T186A and S184A (a MAPKKK consensus site), prepared as dimers with p25, were evaluated as Cdk1 substrates in vitro. Both wild-type p27 and the S184A variant were phosphorylated, but the T186A variant was not (Figure 3C). The p25 component of the p27/p25 heterodimer was not phosphorylated.

p27 phosphorylation and Plk1 binding. (A) Location in mammalian p27 of the predicted Cdk1 phosphorylation site (T186, red; Group-based Prediction System; GPS Version 2.1, http://gps.biocuckoo.org/) and the Plk1 binding motif (red underline). (B) In vitro phosphorylation of native bovine and chick embryo brain dynactin. The autoradiograms (left four lanes) show samples incubated in the absence (−) or presence (+) of purified Cdk1. Arrowheads indicate p27; asterisks mark cyclin B, which is autophosphorylated. The lane on the right shows bovine dynactin (Coomassie blue stain). (C) In vitro phosphorylation by Cdk1 of bacterially expressed GST-p27/p25His6 complexes (wild-type p27 and the T186A and S184A variants) and a GST control. Left: Coomassie blue-stained gels, right: the corresponding autoradiograms. (D) In vivo phosphorylation of endogenous p27. Lysates prepared from Cos7 cells synchronized in G1 (17h after double thymidine block) or M (via nocodazole arrest and shake off) were subjected to two-dimensional immunoblotting for p27. (E) In vivo phosphorylation of exogenously expressed wild-type and T186A EGFP-p27 was evaluated as in (D). (F) In vitro binding of purified p27/p25 heterodimers to GST-tagged wild type PBD (wt) and a non-binding variant (mut) (Elia et al, 2003). Proteins immobilized on glutathione beads were incubated with purified p27/p25 heterodimers that had been phosphorylated in vitro using purified Cdk1. Phosphorylation was verified by ProQ staining. +: phosphorylated p27; −: unphosphorylated p27.
Source data for this figure is available on the online supplementary information page.
To evaluate p27 phosphorylation in vivo, we performed two-dimensional immunoblotting on lysates of Cos7 cells synchronized in G1 or M phase (Figure 3D). p27 was seen to be phosphorylated in M phase, but not in G1. When expressed in cells, E-GFP-tagged wild-type p27 also underwent mitotic phosphorylation, whereas the T186A mutant did not (Figure 3E). Taken together, these results verify that p27 is mitotically phosphorylated at Thr186 in vivo.
Phospho-Thr186 is part of a consensus binding site for the master regulator, Plk1, that acts broadly during the process of cell division, from mitotic entry through cytokinesis. It is found at multiple sites in the spindle, including spindle poles, kinetochores, the spindle midzone and the intracellular cytokinetic bridge, all sites where dynactin and dynein also accumulate. Plk1 is recruited to these sites via a number of different binding partners (Petronczki et al, 2008). Dynactin, more specifically its p27 subunit, may be a previously unidentified Plk1 targeting factor. To demonstrate that p27 can bind Plk1 directly, we performed in vitro experiments using GST-tagged polo-binding domain (PBD; AA 365–603 of human Plk1) and purified recombinant p27/p25 heterodimers that had been phosphorylated by purified Cdk1 in vitro (Figure 3F). Phospho-p27 bound to immobilized wild-type PBD but not a PBD variant that is attenuated for binding (Elia et al, 2003). This interaction was dependent on Cdk1 phosphorylation of p27 because minimal binding was observed to the non-phosphorylated control.
The crystal structure of dynactin p27
To gain further insight into structure–function relationships in the p27/p25 heterodimer and to elucidate the structural context of the p27 phosphorylation site, we embarked on a crystallographic study of these proteins. Amino-acid sequence-based analysis of p27 and p25 using 3D-Jury (Ginalski et al, 2003; Kajan and Rychlewski, 2007) suggested that both proteins contain an unusual left-handed parallel β-helix (LβH) domain, in agreement with earlier hypotheses (Parisi et al, 2004). Co-expression of the two proteins in E. coli using a bi-cistronic vector resulted in a stable, soluble complex that was used in the in vitro phosphorylation and PBD binding experiments described above. Unfortunately, the complex did not yield crystals suitable for structural characterization despite extensive effort. Interestingly, recombinant isolated human p27, but not p25 (which is unstable in the absence of p27), could also be expressed in soluble form in E. coli. Starting with this protein, we obtained a good-quality atomic model using diffraction data to 2.2Å resolution (see Supplementary data, Materials and methods; Table I). The asymmetric unit contains two independent monomers, each with interpretable electron density for residues Ser8 to Thr70 and Lys80 to Gln159, accounting for 74% of the polypeptide chain.
Table 1

aThe numbers in parentheses describe the relevant value for the last resolution shell.
bRsym=∑|Ii−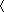 I
I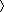 |/∑I, where Ii is the intensity of the ith observation, and I is the mean intensity of the reflections.
|/∑I, where Ii is the intensity of the ith observation, and I is the mean intensity of the reflections.
cRcryst=∑||Fobs|−|Fcalc||/∑|Fobs|, crystallographic R factor, and Rfree=∑||Fobs|−|Fcalc||/∑|Fobs|, where all reflections belong to a test set of randomly selected data.
The ordered domain of p27 visualized by the crystal structure (Figure 4) shows the predicted LβH tertiary fold, making it the first vertebrate protein experimentally shown to display this architecture. Like its right-handed counterpart, this fold contains a β-helical coil formed by repeating units of three β-strands (‘rungs’) separated by turns or loops where the polypeptide chain takes a 300° turn (Choi et al, 2008). The rungs pack against each other by main-chain to main-chain hydrogen bonds parallel to the helical axis. The p27 LβH domain is a type I β-helix, containing seven rungs, with six residues per strand (i.e., 18 residues per rung). The first four amino acids in each rung display the canonical β-sheet secondary structure, with the side chains of the first and third pointing into the interior of the protein, and the side chains of the second and fourth pointing into the solvent. The fifth and sixth residues in each rung make up the turn. Thus, the molecule has a distinctly triangular cross section, with three faces (A, B and C) separated by edges (Figure 4A, referred to as ‘Turns’ in Figure 4C).

The structure of the LβH domain of human p27 (PDB accession number: 3TV0). (A) A view of the crystallographic model of the LβH domain looking down the main axis of the β-helix, from the N-terminus towards the C-terminus. The unique features of the β-helical fold, and residues in the key position 2, are shown in detail using different colours. (B) A view looking directly at Face B of the LβH. The molecular surface is coloured according to element (C: white, N: blue, O: red, and S: yellow). (C) Structure-based amino-acid sequence alignment of the seven ‘rungs’ of the p27 β-helix, coloured to match the structural model in (A). Amino acids that face the interior of the β-helix and thus contribute to the hydrophobic core are indicated in bold. The grey shading indicates the hydrophobic amino acids in Face B (the central ridge in B) that may contribute to the dimer interface.
The three faces of the p27 LβH domain are characterized by different types of exposed amino acids. In each rung, the second residue is positioned in the centre and thus contributes significantly to the physicochemical properties of that respective face. Faces A and C show a variety of amino acids in position 2, but face B has exclusively hydrophobic amino acids at this position (Figure 4C). This is likely to have functional significance, as discussed below. The edge between the A and B faces (Turn 1) is made up of regular, two-residue long turns, as is the edge between the B and C faces (Turn 2). The turns that make up the edge between the C and A faces (Turn 3) are irregular and include two longer loops, one comprising residues 43–47 and a longer one comprising residues 64–81, most of which are not resolved in the electron density map.
Beyond Gln159, the last residue visible in the electron density, secondary structure prediction suggests the presence of a short disordered sequence, a region with high propensity to form an α-helix (residues 170–185; a similar α-helix is predicted in p25), and a disordered C-terminus. The Cdk1 phosphorylation site, Thr186, is the first residue in the disordered segment, suggesting high accessibility to kinase.
A model for p25 structure and mechanism of heterodimerization
The shared predicted LβH tertiary fold of p27 and p25 allowed us to propose a model of the p25 LβH domain based on the p27 crystal structure. The model (Figure 5) shows that the C face of the p25 LβH domain also contains a ridge of exposed hydrophobic residues, making this face structurally similar to the B face of p27. This raises the possibility that the p27 and p25 proteins utilize their respective B and C faces to interact within the heterodimer. This hypothesis is corroborated by the fact that within the asymmetric unit of the p27 crystal, two independent monomers form an intimate homotypic interface between the two B faces, burying ~1300Å2 of solvent exposed surface (Figure 5A). In this geometry (irrespective of orientation), the loops in Turn 3 of both proteins are near each other, raising the possibility that this part of the structure contributes to interactions with other dynactin components or extrinsic binding partners.

A proposed dimerization mode for p27 and p25. (A) The crystallographic p27 homodimer, visualized perpendicular to the dimerization axis with the residues of the hydrophobic ridge on the B face highlighted. (B) Structure-based amino-acid sequence alignment of the seven ‘rungs’ of p25. β-helix faces are coloured as in Figure 4. The grey shading indicates the hydrophobic amino acids in Face C that are predicted to form the dimer interface. Amino acids that face the interior of the β-helix and thus contribute to the hydrophobic core are indicated in bold. (C) A model of the physiological p27/p25 heterodimer. The hydrophobic amino acids exposed on the surfaces of Faces B and C of p27 and p25 (respectively) are depicted with red spheres. The locations of the two edges that contain extended loops (between faces A and C) are also indicated.
Cdk1-phosphorylated p27 targets Plk1 to kinetochores
Our imaging analysis indicates that loss of the p27/p25 heterodimer from kinetochore-associated dynactin allows cells to enter anaphase more quickly without altering steady-state levels of many kinetochore-associated molecules. To explore the possibility that the accelerated mitotic progression might be due to impaired Plk1 targeting to kinetochores, we examined Plk1 localization in mitotic cells. The amount of Plk1 present at spindle poles and in intracellular cytokinetic bridges was unchanged in cells depleted of any of the dynactin subunits (Figure 6A–C) indicating that Plk1 recruitment to these sites depends primarily upon binding partners other than the dynactin p27 component. However, cells from which kinetochore dynactin was removed completely (by p150Glued or Arp11 depletion) or stripped of p27/p25 (by p27 depletion) showed a significant reduction in Plk1 at prometaphase kinetochores (Figure 6D and F). These results indicate that dynactin, specifically its p27 and p25 subunits, is required for full Plk1 targeting to kinetochores. The residual Plk1 present at kinetochores in p27/p25-depleted cells is most likely recruited via other known polo-binding proteins such as Bub1, BubR1, PBIP1 and/or NudC, none of which are reduced as a result of p27/p25 depletion (Figure 2C; Supplementary Figure 4).

Analysis of Plk1 localization to mitotic structures in cells depleted of dynactin subunits. (A) Plk1 fluorescence intensities in cytokinetic bridges (B) and at spindle poles (C) of cells co-stained for tubulin (MT) were quantified, normalized to microtubule pixel values in the same area and expressed as per cent of controls (mean±s.e.m., n>150 cells). The loss of midbody density seen in (B) is commonly seen in cells depleted of dynactin subunits. Bar=5μm. (D through K) Kinetochore localization of Plk1 and 3F3/2. (D, E) Representative images from asynchronous Cos7 populations fixed and stained for Plk1 (green, left panels) or 3F3/2 (green, right panels) plus anti-centromere antigen (ACA; red). Representative prometaphase cells are shown here. (E) Cells stained after brief nocodazole treatment are shown. Bar=5μm. The insets (3 ×) show merged images of Plk1 or 3F3/2 with ACA. (F through K) Quantitative analysis of Plk1 (F, H, J) and 3F3/2 (G, I, K) fluorescence intensity at kinetochores, normalized to ACA pixel values (mean±s.e.m., n>600 kinetochores, 15 cells). (F, G) Cells transfected with different dynactin subunit siRNAs in the presence (+) or absence (−) of the wt p27 rescue vector. (H, I) Cells stained after brief nocodazole treatment. (J, K) Cells cotransfected with control or p27 siRNAs and the rescue vectors indicated (vector=pCAGIG lacking an insert).
Source data for this figure is available on the online supplementary information page.
To verify that dynactin-mediated recruitment of Plk1 to kinetochores was functionally relevant, we used the 3F3/2 antibody to assess the levels of phosphorylated Plk1 substrates. This antibody recognizes a phosphoepitope generated by Plk1 phosphorylation on a number of spindle-associated proteins (Ahonen et al, 2005; Wong and Fang, 2005). Kinetochore labelling with 3F3/2 has been shown to be reduced when Plk1 is depleted or inhibited (Ahonen et al, 2005; Wong and Fang, 2005). 3F3/2 labelling was significantly reduced at kinetochores of cells treated with dynactin subunit siRNAs, verifying that Plk1 activity is attenuated (Figure 6D and G). Depletion of Spindly, which causes both dynein and dynactin to be lost from kinetochores (Griffis et al, 2007; Chan et al, 2009; Barisic et al, 2010; Gassmann et al, 2010) and thus removes p27, also resulted in dramatically reduced Plk1 binding to prometaphase kinetochores (Supplementary Figure 5). The reduction in Plk1 and 3F3/2 staining seen in cells depleted of dynactin subunits reflected the intrinsic composition of the kinetochore because similar results were observed in nocodazole-treated cells in which MT-dependent tension has been eliminated (Figure 6E, H and I).
These findings provide strong evidence that dynactin targets Plk1 to mitotic kinetochores via phospho-Thr186 in the p27/p25 heterodimer. As a final test of this hypothesis, we evaluated whether the non-phosphorylatable p27 variant, T186A, expressed at roughly the same level as endogenous p27 so it incorporates fully into dynactin-like wild-type p27 (Supplementary Figure 1), could rescue the mitotic phenotypes seen in cells depleted of p27 and p25. Wild-type p27, but not the T186A variant, fully rescued the mitotic timing defects (Figure 1A, B and F; Supplementary Movies 4 and 10). It also restored Plk1 and 3F3/2 at kinetochores to normal levels (Figure 6F and G), whereas the T186A variant did not (Figure 6J and K; Supplementary Figure 6). In cells depleted of Arp11 or p150Glued, expression of wild-type p27 did not restore kinetochore Plk1 or 3F3/2 to normal levels (Figure 6F and G), as expected given that p27/p25 is not recruited to kinetochores unless they are incorporated into dynactin (Supplementary Figure 3). Expression of neither wild-type p27 nor the T186A variant had a discernable impact on mitotic timing or Plk1 targeting in cells transfected with control siRNAs (Supplementary Figures 6 and 7), verifying that low level expression of these proteins in cells containing endogenous dynactin has no effect.
Kinetochore–MT attachment, but not kinetochore spacing, is perturbed when p27 and p25 are removed from dynactin
Kinetochore-associated Plk1 has been implicated in the establishment of MT–kinetochore attachments (Sumara et al, 2004; Lenart et al, 2007; Petronczki et al, 2008). To determine whether MT–kinetochore interactions were perturbed in cells depleted of dynactin subunits, we treated cells with monastrol to generate monopolar spindles. These structures allow intimate end-on attachments to be distinguished from lateral interactions and unattached kinetochores (Kapoor et al, 2000; Lenart et al, 2007). Under normal conditions, this assay reveals that >90% of kinetochores sit at the ends of bundles of MTs that emanate from the pole in an orientation where one sister faces the pole the other sister faces away and no MTs appear to run between them (i.e., mono-oriented k-fibres; Figure 7A and B). Mono-oriented k-fibres are seen in only about 60% of cells depleted of p150Glued and we see an increased number of cases where tubulin bundles run between sisters (lateral k-fibres), consistent with an increase in merotelic attachments. In cells depleted of p27/p25 we also observed fewer mono-oriented attachments and more lateral associations. This is as expected given that these cells show an increased number of uncongressed and lagging chromosomes (Figure 1E) and supports the idea that p27/p25-based recruitment of Plk1 contributes to proper establishment of kinetochore–MT interactions.

Effect of dynactin subunit depletion on kinetochore behaviour. (A) Representative images of monopolar spindles generated by monastrol treatment of asynchronous Cos7 cells stained with DAPI plus Abs to tubulin and ACA. Structures were observed using deconvolution microscopy. These images are maximal projections of three z-planes but kinetochore–microtubule associations were assessed by examining the entire deconvolved image. The insets provide images of selected kinetochore pairs at higher magnification to illustrate the different interactions seen. Bar=5μm. (B) Images as in (A) were scored for the types of kinetochore–microtubule interaction observed (end-on=mono-oriented k-fibres; lateral=lateral k-fibres; mean±s.d., n>100 kinetochore pairs per condition). (C) The distances between sister kinetochores on bi-oriented chromosomes were measured in asynchronous Cos7 cells stained with DAPI plus Abs to tubulin and ACA (mean±s.e.m.; *P<0.01 compared to control; N=number of kinetochores). Taxol treatment (1μM, 12min) was used to provide a control for the amount of tension expected at unattached kinetochores. (D, E) Analysis of kinetochore Mad1 in cells treated with dynactin subunit siRNAs. (D) Representative images of Mad1 localization at prometaphase kinetochores in asynchronous cell populations. Bar=5μm. (E) Quantitation of Mad1 levels at kinetochores of unaligned (i.e., prometaphase) and aligned (i.e., metaphase) chromosomes (judged by DAPI staining). Mad1 fluorescence intensity was normalized to ACA pixel values (mean±s.e.m., n>600 kinetochores, 15 cells) and expressed as per cent of the intensity at kinetochores of unaligned chromosomes in control cells. (F, G) As in (D) and (E), but in cells subjected to nocodazole treatment. Bar=5μm.
Source data for this figure is available on the online supplementary information page.
The spindle assembly checkpoint has been proposed to monitor tension between and within kinetochores to ensure that chromosomes are properly bi-oriented before anaphase is initiated. Kinetochores from which dynactin or dynein has been removed exhibit decreased tension between sister kinetochore pairs, as evidenced by closer interkinetochore spacing (Varma et al, 2008; Maresca and Salmon, 2009). Depletion of dynactin from kinetochores via Arp11 or p150Glued siRNA treatment also reduced interkinetochore spacing in bi-oriented pairs (Figure 7C). This is most likely due to the absence of dynein, which is believed to exert poleward pulling forces at kinetochores (Varma et al, 2008). Depletion of p27/p25, by contrast, does not result in a loss of tension, as expected given that kinetochores in these cells still have normal steady-state levels of dynein. This suggests that inhibition of Plk1 binding to dynactin at kinetochores does not alter the tension generating mechanism, but instead interferes with the generation of and/or prematurely terminates the ‘wait-anaphase’ signal.
p27/p25 contributes to recruitment of Mad1 to kinetochores
Kinetochore-associated Plk1 has also been implicated in the recruitment of spindle checkpoint proteins to kinetochores and checkpoint signalling (Ahonen et al, 2005; Kang et al, 2006; Elowe et al, 2007). A key governor of the checkpoint is the MCC. This ‘wait-anaphase’ inhibitory complex, comprising the proteins Mad2, BubR1, Bub3 and Cdc20, is assembled at kinetochores of unaligned chromosomes and is disassembled prior to anaphase in a process that involves component removal (‘stripping’) by the dynein/dynactin motor. Recruitment of the essential MCC component, Mad2, depends upon the presence of a binding partner, Mad1, at kinetochores. Like the MCC, Mad1 is present on the kinetochores of unaligned chromosomes but is transported away by dynein/dynactin once chromosomes are properly aligned and the checkpoint is satisfied (DeLuca et al, 2003; Figure 7E, Control, right). Unlike what was observed in controls, Mad1 levels were high on aligned chromosomes in cells treated with either Arp11 or p150Glued siRNAs. This is expected given that their kinetochores contain less dynein/dynactin that can remove Mad1. Aligned chromosomes in cells depleted of p27, by contrast, showed very low levels of Mad1, just like controls, consistent with the fact that their kinetochores bear a normal amount of dynein. Mad1 levels on the kinetochores of unaligned chromosomes in cells lacking the dynactin-bound pool of Plk1 (i.e., cells treated with p27, Arp11 or p150Glued siRNAs) were significantly lower than controls (Figure 7D and E). A similar result was seen in cells treated with nocodazole (Figure 7F and G) or depleted of Spindly (Supplementary Figure 5). Although it has been reported that Spindly depletion has no effect on Mad1 (or Mad2) localization (Chan et al, 2009; Barisic et al, 2010; Gassmann et al, 2010), efficiency of knockdown or sensitivity of Mad1 recruitment to Spindly levels in the different cell lines used in these studies may explain the discrepancy. In any case, our results indicate that the dynactin p27 component is necessary for binding or retention of Mad1 at kinetochores. This might be due to phosphorylation of Mad1, a known Plk1 substrate (Grosstessner-Hain et al, 2011; Kettenbach et al, 2011; Santamaria et al, 2011), or phosphorylation of a molecule such as dynein that governs Mad1 retention at kinetochores. Reduced levels of Mad1 at unaligned prometaphase kinetochores would interfere with the assembly of the MCC and might contribute to the accelerated mitotic progression we observe.
Discussion
In this study, we show that dynactin’s p27 component contributes to cell-cycle progression in a completely unexpected way. Rather than helping target the dynein/dynactin motor to motile cargoes as it does in interphase, mitotically phosphorylated p27 recruits an extrinsic protein, Plk1, to the dynein motor complex at kinetochores. Our crystallographic study reveals that the Plk1 docking site is located near the C-terminus of p27, in a region outside the well-folded, type I left-handed β-helix domain.
All but one structurally characterized type I LβH family members are bacterial enzymes that typically exhibit acyltransferase activity, but eukaryotic p27 lacks the residues required for catalysis. The only other proteins in the human genome predicted to share the type I LβH fold are the ε and γ subunits of the translation initiation factor eIF2B and GDP-mannose pyrophosphorylase A and B (Choi et al, 2008). The evolutionary origins of these rare eukaryotic LβH proteins are not known and it is unclear how their unusual structure contributes to their physiological functions.
Dynein and dynactin have numerous, well-characterized functions in the mitotic spindle. They are recruited to kinetochores via multiple binding partners, including NudE, NudEL, Spindly and the RZZ complex, which provides for a complicated protein–protein interaction network (Bader and Vaughan, 2010). Loss of any component impairs dynein/dynactin binding (Starr et al, 1998; Griffis et al, 2007; Yang et al, 2007; Chan et al, 2009) and leads to improper kinetochore–MT attachments in prometaphase (Sharp et al, 2000; Gassmann et al, 2008; Varma et al, 2008) and loss of Mad1 and Mad2 (Buffin et al, 2005; Kops et al, 2005). In metaphase, impairment of dynein/dynactin function leads to prolonged checkpoint activation and retention of kinetochore-associated checkpoint proteins, which has led to the hypothesis that dynein-based ‘stripping’ of key regulators from kinetochores drives checkpoint silencing (Howell et al, 2000; Sivaram et al, 2009). Dynein/dynactin perturbation also typically results in significant spindle derangement owing to the focusing activities they provide at spindle poles, which makes it all the more difficult to tease out their roles at kinetochores. Because p27/p25 depletion does not affect spindle formation or dynein/dynactin integrity and recruitment to kinetochores, but does result in selective loss of Plk1 from this site, it represents a powerful new tool for analysing the functions of kinetochore-associated Plk1 and its downstream targets in the processes of chromosome congression and execution of the spindle assembly checkpoint.
Kinetochore-associated Plk1 has been implicated in kinetochore–MT engagement and checkpoint protein recruitment (Sumara et al, 2004; Ahonen et al, 2005; Wong and Fang, 2005; Elowe et al, 2007; Lenart et al, 2007; Hood et al, 2012; Liu et al, 2012; Maia et al, 2012). Our data suggest that full Plk1 activity is needed for establishment of proper kinetochore–MT attachments. However, unlike what is usually observed when kinetochore–MT interactions are impaired, cells depleted of p27 do not exhibit prolonged spindle assembly checkpoint activation, delay of anaphase onset and mitotic arrest. Instead, the checkpoint, if it is activated at all, appears to be prematurely satisfied, resulting in accelerated mitotic progression. Our findings suggest the following scenarios, which are not mutually exclusive. p27-based recruitment of Plk1 to kinetochores may contribute to recognition or resolution of merotelic attachments, or it may influence recruitment, activation and/or retention of checkpoint proteins.
Impaired resolution of merotelic attachments in cells lacking p27 and p25 could lead to accelerated mitotic progression if this is a predominant mode of attachment, because such a situation would not activate the spindle assembly checkpoint. Selective pharmacological perturbation of kinetochore Plk1 has been shown to yield an increase in merotelic attachments (Lera and Burkard, 2012). Three major contributors to resolution of such attachments, Aurora B, MCAK and Kif2B, are known Pkl1 substrates (Grosstessner-Hain et al, 2011; Kettenbach et al, 2011; Santamaria et al, 2011; Hood et al, 2012).
Accelerated mitotic progression might also be the result of aberrant checkpoint protein turnover. Unaligned chromosomes in cells depleted of p27/p25 show greatly reduced levels of Mad1, a protein that plays a key role in assembly of the MCC. Plk1 has been reported to phosphorylate the MCC component BubR1, as well as Mad1, which binds the MCC component Mad2 and facilitates its incorporation into the complex (Elowe et al, 2007; Grosstessner-Hain et al, 2011; Kettenbach et al, 2011; Santamaria et al, 2011). Phosphorylation might alter the affinity of these proteins for their binding partners, enhancing MCC formation and/or retention at the kinetochore. Plk1 has also been linked to phosphorylation of dynein on its intermediate and light intermediate chains (Bader et al, 2011; Grosstessner-Hain et al, 2011; Santamaria et al, 2011). Because dynactin binds dynein directly, it provides an ideal mechanism for localizing Plk1 activity nearby to regulate dynein activity. Plk1 phosphorylation is thought to reduce dynein’s affinity for dynactin. This has been proposed to prevent the motor from switching from a state in which it is inactive for motility and tightly bound to the RZZ complex to a fully active form that can remove checkpoint proteins (Bader et al, 2011). Attenuation of a Plk1-dependent switch might yield constitutively or prematurely motile dynein, which has the potential to impact the checkpoint in multiple ways. Dynein-based removal of Mad1 would prevent conversion of Mad2 from its autoinhibited form to the form that is incorporated into the MCC (Fava et al, 2011). Alternatively, constitutively active dynein might prematurely remove the fully assembled MCC, triggering anaphase. Finally, enhanced dynein activity would be expected to generate more tension at the kinetochore and increase intrakinetochore stretch which has been correlated with checkpoint satisfaction (Maresca and Salmon, 2009).
Materials and methods
Cell culture, RNAi, 2D immunoblotting, immunofluorescence and image analysis
See Supplementary data for details.
Antibodies
Commercial antibodies are α-tubulin (DM1A), phospho-histone 3 (serine 10): Sigma; p150Glued (clone 1): BD, DIC clone 74.1: Millipore; p25 and p27: ProteinTech Group; Plk1: Santa Cruz; GFP: Invitrogen; PBIP1: Rockland; NudC: Abcam; GST: GE, Alexa488-, Alexa568-, and Alexa633-conjugated secondary antibodies: Invitrogen; alkaline phosphatase-conjugated secondary antibodies: Tropix. Monoclonal antibodies against p27 (27A) and Arp1 (45A) were produced and purified as described previously (Schafer et al, 1994; Melkonian et al, 2007). Other antibodies were obtained from our colleagues: mouse 3F3/2 ascites (Campbell and Gorbsky, 1995): Gary Gorbsky; rat anti-ZW10 3.2 (Chan et al, 2000): Gordon Chan, sheep anti-Bub1 (SB1.3), BubR1 (SBR1.1), Bub3 (SB3.2), MPS1 (SMP1.1) (Taylor et al, 2001; Tighe et al, 2008; Gurden et al, 2010): Stephen Taylor; rabbit anti-Spindly (Griffis et al, 2007): Eric Griffis; mouse anti-Mad1 (BB3–8) (De Antoni et al, 2005): Andrea Musacchio; human anti-centromere serum (ACA; Martin et al, 1990): JB Rattner.
Dynactin purification
Chicken embryo and bovine brain dynactin were purified as described previously (Schroer and Sheetz, 1991; Bingham et al, 1998).
Expression and purification of p25/p27 heterodimers
Full-length human p27 and full-length mouse p25 cDNA (ATCC) were amplified and cloned into pDEST15 to create the bi-cistronic expression vector pDEST-GST-p27/p25-His6, with an rTEV cleavage site downstream of GST and a ribosomal binding site (RBS) between the p27 and p25 coding sequences. The non-cleavable C-terminal His6 tag was inserted for purification purposes. The T186A (ACT to GCT) and S184A (AGC to GCC) p27 variants were created in pDEST-GST-p27/p25-His6 via QuikChange. BL21-CodonPlus (DE3)-RIL cells (Agilent) were transformed with the plasmid and grown to an OD600 of 0.6 in 0.5l LB at 37°C, then induced with 0.1mM IPTG for 16h at 18°C. For purification of GST-tagged heterodimers, the bacterial pellets were resuspended in 40ml PBS (pH 7.4) containing 1mM DTT and 1mg/ml lysozyme, rotated for 20min at 25°C and sonicated. The clarified bacterial supernatants were applied to a GSTtrap FPLC column (GE) and eluted with 50mM Tris–HCl, 100mM NaCl, 10mM L-glutathione and 1mM DTT, pH 8.0.
GST-PBD binding assays
Preparation of GST-PBD beads: The human Plk1 PBD (AA 365–603; accession #BC01486) from a pCMV-SPORT6 clone (ATCC) was cloned into pGEX4T-2. The PBD binding-null variant, GST-PBD H538A/K540M (Elia et al, 2003), was generated using engineered PCR primers that also contained a unique restriction site (BsaBI) for insertion into the parent vector. The constructs were transformed into BL21-CodonPlus (DE3)-RIL, grown to an OD600 of 0.6 at 37°C in 0.5l LB and induced with 0.1mM IPTG for 3h at 30°C. The bacterial pellet was resuspended in 40ml binding buffer (20mM Tris–HCl, 250mM NaCl, 1mM EDTA, 1mM NaVO4, 20mM NaF, 5mM β-ME, pH 8.0) containing 1mg/ml lysozyme, 5U/ml Benzonase and protease inhibitors, subjected to a freeze/thaw cycle, and sonicated. Two milliliters of the post-sonication supernatant was incubated with Glutathione-Sepharose (for each 10μl aliquot; GE) pre-blocked with 5% non-fat milk in binding buffer, washed 5 × with 500μl binding buffer, then test proteins were added.
To prepare p27/p25 heterodimers for the binding assay, the GST tag was cleaved via TEV protease (Invitrogen) for 16h at 4°C from samples that had been phosphorylated in vitro (see below). The p27/p25 heterodimers were separated from GST and TEV protease using a Superose12 FPLC column attached to a GSTrap column (GE) in 20mM Tris–HCl, 250mM NaCl, 1mM EDTA, 1mM NaVO4, 20mM NaF, 5mM β-ME, pH 8.0. The peak fractions containing p27/p25 heterodimers were identified by Coomassie staining. The purified p27/p25 heterodimers (25μg) were incubated with 10μl Glutathione-Sepharose carrying GST-PBD for 1h at 4°C. The beads were washed with 3 × 500μl binding buffer and proteins were eluted in 500μl binding buffer plus 30mM L-glutathione for 1h at 4°C prior to analysis of 10μl by immunoblotting.
In vitro phosphorylation
To detect Cdk1 phosphorylation, purified dynactin (27nM) or recombinant GST-p27/p25-His6 heterodimers (0.57μM) were incubated with 10 units of Cdk1 (0.01μg, NEB), 100μM ATP and 3.75 μCi γ-32P ATP in Cdk1 buffer in a 15-μl reaction volume for 30min at 30°C. The entire sample was subjected to SDS–PAGE and autoradiography.
To generate phosphorylated proteins for GST-PBD binding, 350μg purified GST-p27/p25-His6 heterodimers were incubated with 20 units of Cdk1 (NEB) plus 500μM ATP in a 1-ml volume for 16h at 30°C. A parallel sample incubated without Cdk1 was used as the non-phosphorylated control. Phosphorylation was verified using Pro-Q Diamond Stain (Invitrogen).
Live-cell imaging
To image mitosis in cells expressing histone 2B-mCherry, cells were harvested by trypsinization 24h after transfection with siRNAs, histone 2B-mCherry and rescue plasmids, then 1.5 × 105 cells were plated into glass bottom dishes (MatTek) and cultured in HEPES-buffered DMEM for 24h prior to synchronization using a double thymidine block. Live-cell recordings were initiated at 9–10h after release from the block (G1/S boundary). Cells were imaged on a Zeiss Axiovert 200M inverted microscope (40 × /0.85 NA Plan-Neofluor air objective) heated to 37°C using an In Vivo Scientific incubator. Images were collected using a Cooke SensiCam camera. Samples were illuminated at 25% intensity via a 120W X-Cite 120PC Xenon arc lamp (EXFO Life Science). To reduce intensity and prevent radiation-induced changes in the cell cycle (Khodjakov and Rieder, 1999), the light beam was passed through GG400 (UV) and KG5 (IR) filters (Newport Corporation) as well as a 12% neutral density filter (Zeiss). This filter combination allowed cells to proceed through mitosis without discernable delays. Images (100ms/frame) were collected at 120-s intervals. Three z planes (0.5μm step size) were collected at each time point and at the end of the imaging interval the best optical plane for each cell was chosen and used to generate movies in SlideBook.
For phase contrast imaging, RNAi-treated cells synchronized as above were illuminated using a 12V/100W halogen lamp at 30–35% intensity with heat absorbing (KG1) and green interference filters (546±20nm bandpass, Zeiss) (Wadsworth et al, 2005). Time-lapse movies were acquired at 90-s intervals on a Zeiss Axiovert 200 Microscope (LD Plan-Neuofluor 40 × , 0.6 NA Phase2 objective). Three z planes (0.5μm step size) were collected at each time point, and the best optical plane for each cell was used to generate movies.
Analysis of interkinetochore spacing and kinetochore–MT interactions
Cells transfected with siRNAs were methanol fixed, stained for tubulin and ACA, and 0.15μm image planes were collected as for immunofluorescence. Image stacks were processed using SlideBook Constrained Iterative deconvolution software and deconvolved images were exported to Image J. Only bi-oriented kinetochore pairs were used for measurement of the interkinetochore distance.
Kinetochore–MT interactions were analysed in cells treated with 100μM monastrol (Sigma) for 2h followed by tubulin and ACA immunostaining, and deconvolution imaging as described (Lenart et al, 2007).
Supplementary Material
Acknowledgments
We thank Ms Natalya Olekhnovich for technical assistance. We are grateful to Drs G Chan, G Gorbsky, E Griffis, JB Rattner, A Musacchio and S Taylor for generously providing antibodies, to Dr Q Vong and Y Zheng for the histone 2B-mCherry plasmid, and the two anonymous reviewers plus Dr T Stukenberg for input and comments on the manuscript. The use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract No. W-31-109-Eng-38. We thank the SER-CAT staff for assistance with data collection. Supporting institutions of SER-CAT may be found at http://www.ser-cat.org/members.html. This research was supported by grants from the NIH to TAS (GM044589) and ZSD (NS036267) and training grant T32 GM07231 (Johns Hopkins University CMDB Program). Microscopy was performed at the Johns Hopkins Integrated Imaging Center.
Author contributions: TAS and ZSD supervised the experimental design and wrote the paper. TY performed the experiments in Figures 1, ,2,2, 3D, E, ,66 and and7,7, Supplementary Movies, and Supplementary Figures. BRS performed the experiments in Figure 3C and F. FKC performed the experiments in Figure 3B and C. AK, MYZ and UD performed the experiments in Figures 4 and and5.5. All the authors understand their responsibilities connected to authorship.
References
- Ahonen LJ, Kallio MJ, Daum JR, Bolton M, Manke IA, Yaffe MB, Stukenberg PT, Gorbsky GJ (2005) Polo-like kinase 1 creates the tension-sensing 3F3/2 phosphoepitope and modulates the association of spindle-checkpoint proteins at kinetochores. Curr Biol 15: 1078–1089 [Abstract] [Google Scholar]
- Bader JR, Kasuboski JM, Winding M, Vaughan PS, Hinchcliffe EH, Vaughan KT (2011) Polo-like kinase1 is required for recruitment of dynein to kinetochores during mitosis. J Biol Chem 286: 20769–20777 [Europe PMC free article] [Abstract] [Google Scholar]
- Bader JR, Vaughan KT (2010) Dynein at the kinetochore: Timing, Interactions and Functions. Semin Cell Dev Biol 21: 269–275 [Europe PMC free article] [Abstract] [Google Scholar]
- Barisic M, Sohm B, Mikolcevic P, Wandke C, Rauch V, Ringer T, Hess M, Bonn G, Geley S (2010) Spindly/CCDC99 is required for efficient chromosome congression and mitotic checkpoint regulation. Mol Biol Cell 21: 1968–1981 [Europe PMC free article] [Abstract] [Google Scholar]
- Bingham JB, King SJ, Schroer TA (1998) Purification of dynactin and dynein from brain tissue. Methods Enzymol 298: 171–184 [Abstract] [Google Scholar]
- Buffin E, Lefebvre C, Huang J, Gagou ME, Karess RE (2005) Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr Biol 15: 856–861 [Abstract] [Google Scholar]
- Campbell MS, Gorbsky GJ (1995) Microinjection of mitotic cells with the 3F3/2 anti-phosphoepitope antibody delays the onset of anaphase. J Cell Biol 129: 1195–1204 [Europe PMC free article] [Abstract] [Google Scholar]
- Chan GK, Jablonski SA, Starr DA, Goldberg ML, Yen TJ (2000) Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat Cell Biol 2: 944–947 [Abstract] [Google Scholar]
- Chan YW, Fava LL, Uldschmid A, Schmitz MH, Gerlich DW, Nigg EA, Santamaria A (2009) Mitotic control of kinetochore-associated dynein and spindle orientation by human Spindly. J Cell Biol 185: 859–874 [Europe PMC free article] [Abstract] [Google Scholar]
- Choi JH, Govaerts C, May BC, Cohen FE (2008) Analysis of the sequence and structural features of the left-handed beta-helical fold. Proteins 73: 150–160 [Abstract] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, Musacchio A (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15: 214–225 [Abstract] [Google Scholar]
- DeLuca JG, Howell BJ, Canman JC, Hickey JM, Fang G, Salmon ED (2003) Nuf2 and Hec1 are required for retention of the checkpoint proteins Mad1 and Mad2 to kinetochores. Curr Biol 13: 2103–2109 [Abstract] [Google Scholar]
- Echeverri CJ, Paschal BM, Vaughan KT, Vallee RB (1996) Molecular characterization of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J Cell Biol 132: 617–633 [Europe PMC free article] [Abstract] [Google Scholar]
- Eckley DM, Gill SR, Melkonian KA, Bingham JB, Goodson HV, Heuser JE, Schroer TA (1999) Analysis of dynactin subcomplexes reveals a novel actin-related protein associated with the Arp1 minifilament pointed end. J Cell Biol 147: 307–320 [Europe PMC free article] [Abstract] [Google Scholar]
- Elia AE, Cantley LC, Yaffe MB (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299: 1228–1231 [Abstract] [Google Scholar]
- Elowe S, Hummer S, Uldschmid A, Li X, Nigg EA (2007) Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev 21: 2205–2219 [Europe PMC free article] [Abstract] [Google Scholar]
- Fava LL, Kaulich M, Nigg EA, Santamaria A (2011) Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J 30: 3322–3336 [Europe PMC free article] [Abstract] [Google Scholar]
- Gassmann R, Essex A, Hu JS, Maddox PS, Motegi F, Sugimoto A, O'Rourke SM, Bowerman B, McLeod I, Yates JR 3rd, Oegema K, Cheeseman IM, Desai A (2008) A new mechanism controlling kinetochore-microtubule interactions revealed by comparison of two dynein-targeting components: SPDL-1 and the Rod/Zwilch/Zw10 complex. Genes Dev 22: 2385–2399 [Europe PMC free article] [Abstract] [Google Scholar]
- Gassmann R, Holland AJ, Varma D, Wan X, Civril F, Cleveland DW, Oegema K, Salmon ED, Desai A (2010) Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev 24: 957–971 [Europe PMC free article] [Abstract] [Google Scholar]
- Ginalski K, Elofsson A, Fischer D, Rychlewski L (2003) 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics 19: 1015–1018 [Abstract] [Google Scholar]
- Griffis ER, Stuurman N, Vale RD (2007) Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J Cell Biol 177: 1005–1015 [Europe PMC free article] [Abstract] [Google Scholar]
- Grosstessner-Hain K, Hegemann B, Novatchkova M, Rameseder J, Joughin BA, Hudecz O, Roitinger E, Pichler P, Kraut N, Yaffe MB, Peters JM, Mechtler K (2011) Quantitative phospho-proteomics to investigate the polo-like kinase 1-dependent phospho-proteome. Mol Cell Proteomics 10: M111 008540 [Europe PMC free article] [Abstract] [Google Scholar]
- Gurden MD, Holland AJ, van Zon W, Tighe A, Vergnolle MA, Andres DA, Spielmann HP, Malumbres M, Wolthuis RM, Cleveland DW, Taylor SS (2010) Cdc20 is required for the post-anaphase, KEN-dependent degradation of centromere protein F. J Cell Sci 123: 321–330 [Europe PMC free article] [Abstract] [Google Scholar]
- Hood EA, Kettenbach AN, Gerber SA, Compton DA (2012) Plk1 regulates the kinesin-13 protein Kif2b to promote faithful chromosome segregation. Mol Biol Cell 23: 2264–2274 [Europe PMC free article] [Abstract] [Google Scholar]
- Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED (2000) Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol 150: 1233–1250 [Europe PMC free article] [Abstract] [Google Scholar]
- Kajan L, Rychlewski L (2007) Evaluation of 3D-Jury on CASP7 models. BMC Bioinformatics 8: 304. [Europe PMC free article] [Abstract] [Google Scholar]
- Kang YH, Park JE, Yu LR, Soung NK, Yun SM, Bang JK, Seong YS, Yu H, Garfield S, Veenstra TD, Lee KS (2006) Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol Cell 24: 409–422 [Abstract] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ (2000) Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol 150: 975–988 [Europe PMC free article] [Abstract] [Google Scholar]
- Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA (2011) Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 4: rs5. [Europe PMC free article] [Abstract] [Google Scholar]
- Khodjakov A, Rieder CL (1999) The sudden recruitment of γ-tubulin to the centrosome at the onset of mitosis and its dynamic exchange throughout the cell cycle, do not required microtubules. J Cell Biol 146: 585–596 [Europe PMC free article] [Abstract] [Google Scholar]
- Kops GJ, Kim Y, Weaver BA, Mao Y, McLeod I, Yates JR 3rd, Tagaya M, Cleveland DW (2005) ZW10 links mitotic checkpoint signaling to the structural kinetochore. J Cell Biol 169: 49–60 [Europe PMC free article] [Abstract] [Google Scholar]
- Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM (2007) The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 17: 304–315 [Abstract] [Google Scholar]
- Lera RF, Burkard ME (2012) High mitotic activity of Polo-like kinase 1 is required for chromosome segregation and genomic integrity in human epithelial cells. J Biol Chem 287: 42812–42825 [Europe PMC free article] [Abstract] [Google Scholar]
- Li H, Liu XS, Yang X, Song B, Wang Y, Liu X (2010) Polo-like kinase 1 phosphorylation of p150Glued facilitates nuclear envelope breakdown during prophase. Proc Natl Acad Sci USA 107: 14633–14638 [Europe PMC free article] [Abstract] [Google Scholar]
- Liang Y, Yu W, Li Y, Yu L, Zhang Q, Wang F, Yang Z, Du J, Huang Q, Yao X, Zhu X (2007) Nudel modulates kinetochore association and function of cytoplasmic dynein in M phase. Mol Biol Cell 18: 2656–2666 [Europe PMC free article] [Abstract] [Google Scholar]
- Liu D, Davydenko O, Lampson MA (2012) Polo-like kinase-1 regulates kinetochore-microtubule dynamics and spindle checkpoint silencing. J Cell Biol 198: 491–499 [Europe PMC free article] [Abstract] [Google Scholar]
- Liu ST, Rattner JB, Jablonski SA, Yen TJ (2006) Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J Cell Biol 175: 41–53 [Europe PMC free article] [Abstract] [Google Scholar]
- Lowery DM, Clauser KR, Hjerrild M, Lim D, Alexander J, Kishi K, Ong SE, Gammeltoft S, Carr SA, Yaffe MB (2007) Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J 26: 2262–2273 [Europe PMC free article] [Abstract] [Google Scholar]
- Maia AR, Garcia Z, Kabeche L, Barisic M, Maffini S, Macedo-Ribeiro S, Cheeseman IM, Compton DA, Kaverina I, Maiato H (2012) Cdk1 and Plk1 mediate a CLASP2 phospho-switch that stablizes kinetochore-microtubule attachment. J Cell Biol 199: 285–301 [Europe PMC free article] [Abstract] [Google Scholar]
- Maresca TJ, Salmon ED (2009) Intrakinetochore stretch is associated with changes in kinetochore phosphorylation and spindle assembly checkpoint activity. J Cell Biol 184: 373–381 [Europe PMC free article] [Abstract] [Google Scholar]
- Martin L, Cusano R, Kingwell B, Fritzler MJ, Rattner JB (1990) Autoantibodies to chromosomal domains in rheumatic diseases. J Clin Lab Immunol 32: 73–78 [Abstract] [Google Scholar]
- Melkonian KA, Maier KC, Godfrey JE, Rodgers M, Schroer TA (2007) Mechanism of dynamitin-mediated disruption of dynactin. J Biol Chem 282: 19355–19364 [Abstract] [Google Scholar]
- Parisi G, Fornasari MS, Echave J (2004) Dynactins p25 and p27 are predicted to adopt the LbetaH fold. FEBS Lett 562: 1–4 [Abstract] [Google Scholar]
- Petronczki M, Lenart P, Peters JM (2008) Polo on the rise-from mitotic entry to cytokinesis with Plk1. Dev Cell 14: 646–659 [Abstract] [Google Scholar]
- Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, Schmidt A, Sillje HH, Korner R, Nigg EA (2011) The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics 10: M110 004457 [Europe PMC free article] [Abstract] [Google Scholar]
- Schafer DA, Gill SR, Cooper JA, Heuser JE, Schroer TA (1994) Ultrastructural analysis of the dynactin complex: an actin-related protein is a component of a filament that resembles F-actin. J Cell Biol 126: 403–412 [Europe PMC free article] [Abstract] [Google Scholar]
- Schroer T, Cheong FK-Y (2012) Role of dynactin in dynein-mediated motility. In Dyneins: Structure, Biology and Disease King SM (ed). Oxford: Academic Press, , pp. 505–522 [Google Scholar]
- Schroer TA, Sheetz MP (1991) Two activators of microtubule-based vesicle transport. J Cell Biol 115: 1309–1318 [Europe PMC free article] [Abstract] [Google Scholar]
- Sharp DJ, Rogers GC, Scholey JM (2000) Cytoplasmic dynein is required for poleward chromosome movement during mitosis in Drosophila embryos. Nat Cell Biol 2: 922–930 [Abstract] [Google Scholar]
- Sivaram MV, Wadzinski TL, Redick SD, Manna T, Doxsey SJ (2009) Dynein light intermediate chain 1 is required for progress through the spindle assembly checkpoint. EMBO J 28: 902–914 [Europe PMC free article] [Abstract] [Google Scholar]
- Starr DA, Williams BC, Hays TS, Goldberg ML (1998) ZW10 helps recruit dynactin and dynein to the kinetochore. J Cell Biol 142: 763–774 [Europe PMC free article] [Abstract] [Google Scholar]
- Sumara I, Gimenez-Abian JF, Gerlich D, Hirota T, Kraft C, de la Torre C, Ellenberg J, Peters JM (2004) Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol 14: 1712–1722 [Abstract] [Google Scholar]
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ (2001) Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci 114: 4385–4395 [Abstract] [Google Scholar]
- Tighe A, Staples O, Taylor S (2008) Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol 181: 893–901 [Europe PMC free article] [Abstract] [Google Scholar]
- Varma D, Monzo P, Stehman SA, Vallee RB (2008) Direct role of dynein motor in stable kinetochore-microtubule attachment, orientation, and alignment. J Cell Biol 182: 1045–1054 [Europe PMC free article] [Abstract] [Google Scholar]
- Wadsworth P, Rusan NM, Tulu US, Fagerstrom C (2005) Stable expression of fluorescently tagged proteins for studies of mitosis in mammalian cells. Nat Methods 2: 981–987 [Abstract] [Google Scholar]
- Whyte J, Bader JR, Tauhata SB, Raycroft M, Hornick J, Pfister KK, Lane WS, Chan GK, Hinchcliffe EH, Vaughan PS, Vaughan KT (2008) Phosphorylation regulates targeting of cytoplasmic dynein to kinetochores during mitosis. J Cell Biol 183: 819–834 [Europe PMC free article] [Abstract] [Google Scholar]
- Wong OK, Fang G (2005) Plx1 is the 3F3/2 kinase responsible for targeting spindle checkpoint proteins to kinetochores. J Cell Biol 170: 709–719 [Europe PMC free article] [Abstract] [Google Scholar]
- Yang Z, Tulu US, Wadsworth P, Rieder CL (2007) Kinetochore dynein is required for chromosome motion and congression independent of the spindle checkpoint. Curr Biol 17: 973–980 [Europe PMC free article] [Abstract] [Google Scholar]
- Yeh T-Y, Quintyne NJ, Scipioni BR, Eckley DM, Schroer TA (2012) Dynactin's pointed end complex is a cargo-targeting module. Mol Biol Cell 23: 3827–3837 [Europe PMC free article] [Abstract] [Google Scholar]
- Zhang J, Yao X, Fischer L, Abenza JF, Penalva MA, Xiang X (2011) The p25 subunit of the dynactin complex is required for dynein-early endosome interaction. J Cell Biol 193: 1245–1255 [Europe PMC free article] [Abstract] [Google Scholar]
Articles from The EMBO Journal are provided here courtesy of Nature Publishing Group
Full text links
Read article at publisher's site: https://doi.org/10.1038/emboj.2013.30
Read article for free, from open access legal sources, via Unpaywall:
https://europepmc.org/articles/pmc3616283?pdf=render

Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Article citations
Genetic enhancers of partial PLK1 inhibition reveal hypersensitivity to kinetochore perturbations.
PLoS Genet, 19(8):e1010903, 28 Aug 2023
Cited by: 1 article | PMID: 37639469 | PMCID: PMC10491399
Recent Progress on the Localization of PLK1 to the Kinetochore and Its Role in Mitosis.
Int J Mol Sci, 23(9):5252, 08 May 2022
Cited by: 4 articles | PMID: 35563642 | PMCID: PMC9102930
Review
 Free full text in Europe PMC
Free full text in Europe PMCThe Role of Mitotic Kinases and the RZZ Complex in Kinetochore-Microtubule Attachments: Doing the Right Link.
Front Cell Dev Biol, 10:787294, 28 Jan 2022
Cited by: 9 articles | PMID: 35155423 | PMCID: PMC8832123
Review
Free full text in Europe PMCCryo-EM reveals the complex architecture of dynactin's shoulder region and pointed end.
EMBO J, 40(8):e106164, 18 Mar 2021
Cited by: 16 articles | PMID: 33734450 | PMCID: PMC8047447
BUB1 and CENP-U, Primed by CDK1, Are the Main PLK1 Kinetochore Receptors in Mitosis.
Mol Cell, 81(1):67-87.e9, 27 Nov 2020
Cited by: 46 articles | PMID: 33248027 | PMCID: PMC7837267
Go to all (31) article citations
Other citations
Data
Data behind the article
This data has been text mined from the article, or deposited into data resources.
BioStudies: supplemental material and supporting data
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
Polo-like kinase1 is required for recruitment of dynein to kinetochores during mitosis.
J Biol Chem, 286(23):20769-20777, 20 Apr 2011
Cited by: 20 articles | PMID: 21507953 | PMCID: PMC3121523
Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore-microtubule attachments.
EMBO J, 29(17):2953-2965, 27 Jul 2010
Cited by: 60 articles | PMID: 20664522 | PMCID: PMC2944045
Polo-like kinase 1 phosphorylation of p150Glued facilitates nuclear envelope breakdown during prophase.
Proc Natl Acad Sci U S A, 107(33):14633-14638, 02 Aug 2010
Cited by: 34 articles | PMID: 20679239 | PMCID: PMC2930408
Recent Progress on the Localization of PLK1 to the Kinetochore and Its Role in Mitosis.
Int J Mol Sci, 23(9):5252, 08 May 2022
Cited by: 4 articles | PMID: 35563642 | PMCID: PMC9102930
Review
Free full text in Europe PMC
Funding
Funders who supported this work.
NCI NIH HHS (1)
Grant ID: P30 CA044579
NIGMS NIH HHS (5)
Grant ID: R56 GM044589
Grant ID: T32 GM007231
Grant ID: T32 GM07231
Grant ID: GM044589
Grant ID: R01 GM044589
NINDS NIH HHS (2)
Grant ID: NS036267
Grant ID: R01 NS036267



{kind=link}
{kind=link}
{kind=link}