CisOrtho: a program pipeline for genome-wide identification of transcription factor target genes using phylogenetic footprinting.
Review![]() Free full text in Europe PMC
Free full text in Europe PMC
Abstract
Background
All known genomes code for a large number of transcription factors. It is important to develop methods that will reveal how these transcription factors act on a genome wide level, that is, through what target genes they exert their function.Results
We describe here a program pipeline aimed at identifying transcription factor target genes in whole genomes. Starting from a consensus binding site, represented as a weight matrix, potential sites in a pre-filtered genome are identified and then further filtered by assessing conservation of the putative site in the genome of a related species, a process called phylogenetic footprinting. CisOrtho has been successfully used to identify targets for two homeodomain transcription factors in the genomes of the nematodes Caenorhabditis elegans and Caenorhabditis briggsae.Conclusions
CisOrtho will identify targets of other nematode transcription factors whose DNA binding specificity is known and can be easily adapted to search other genomes for transcription factor targets.Free full text
CisOrtho: A program pipeline for genome-wide identification of transcription factor target genes using phylogenetic footprinting
 1
1Abstract
Background
All known genomes code for a large number of transcription factors. It is important to develop methods that will reveal how these transcription factors act on a genome wide level, that is, through what target genes they exert their function.
Results
We describe here a program pipeline aimed at identifying transcription factor target genes in whole genomes. Starting from a consensus binding site, represented as a weight matrix, potential sites in a pre-filtered genome are identified and then further filtered by assessing conservation of the putative site in the genome of a related species, a process called phylogenetic footprinting. CisOrtho has been successfully used to identify targets for two homeodomain transcription factors in the genomes of the nematodes Caenorhabditis elegans and Caenorhabditis briggsae.
Conclusions
CisOrtho will identify targets of other nematode transcription factors whose DNA binding specificity is known and can be easily adapted to search other genomes for transcription factor targets.
Background
Transcription factors are among the most common regulatory proteins in a cell. They subserve a variety of functions during development and homeostasis, yet in most cases the full spectrum of target genes regulated by a given transcription factor is unknown. The identification of transcription factor target genes is a challenging task since most transcription factors have the inherent capacity to tolerate a significant amount of variation in their cis-regulatory binding sites [1]. These variations, which are usually experimentally determined, are commonly represented by a weight matrix with which genomes can be searched [2-4]. However, the usually sparse and widely varying data on transcription factor binding sites together with the large search space of a genome leads to many false hits, thus necessitating further filtering. This can be done by imposing the criteria of conservation of the cis-regulatory sequence in a related species that has diverged long enough ago to cause only functionally relevant DNA sequences to be conserved ("phylogenetic footprinting") [1,5-13]. We describe here a program pipeline that will identify targets of a transcription factor with a defined cis-regulatory target specificity, using two invertebrate model system genomes, those of C. elegans and C. briggsae [14,15].
Implementation
CisOrtho is written in ANSI C++ with the use of the SGI Standard Template Library http://www.sgi.com/tech/stl/ and a supporting freely available options-parsing interface Opt-3.19 (http://nis-www.lanl.gov/~jt/Software/ or from http://dev.wormbase.org/CisOrtho). It consists of 1541 lines of code in 11 source files. The program has two parameters which affect either size (memory) or runtime: Ngenes, number of ortholog-matched gene pairs to consider; D, number of total hits per gene to be reported. Memory is approximately Ngenes* D* 200 bytes; runtime is approximately linear in Ngenes, D and the length of DNA to be searched. For exhaustive settings (Ngenes = 12000, D = 10), total memory is 23.2 MB and runtime 17 minutes on a 2.8 GHz Xeon processor. A more typical run (Ngenes = 500, D = 3) takes 3.9 MB and 42 seconds. Since memory requirement does not depend on the length of DNA to be searched, a search on the Human and Mouse genome would be feasible on a machine with 256 MB.
Results and discussion
Procedure overview
The procedure presented here is aimed at finding genes likely to be regulated by a given transcription factor for which a collection of binding sites is available. There are four steps (Fig. (Fig.1).1). In the first step, annotation files (GFF files) are used to define, classify, and associate with genes, every non-exonic region in the C. elegans and C. briggsae genomes. The second step consists of building a position weight matrix for the set of aligned binding sites, and using it as a scanning window to search the non-exonic regions for the N highest scoring hits, where N is defined by the user. In the third step, we use an available file that provides a one-to-one C. elegans/C. briggsae ortholog-mapping [15] to filter out all ortholog pairs that do not have high-scoring hits for both ortholog members. The fourth step involves sorting the remaining hit-pairs according to the scores of the hits and the number of mismatches between the hits, and finally outputting this in HTML tables. The whole procedure can be conducted at a user-friendly web interface at http://dev.wormbase.org/CisOrtho. A screenshot is shown in Fig. Fig.2.2. It is also available for download at the same website.

Flow chart of program pipeline. Information is shown as rectangles, procedures as ovals. The only user defined inputs are the Transcription Factor Binding Site Alignment file and the number of hits to retrieve. All other input files are downloaded from sources mentioned in the text.

Screenshot of the Web Interface. The address is: http://dev.wormbase.org/CisOrtho. The program will be eventually run by WormBase at http://www.wormbase.org.
Identification of non-exonic regions
We classified all non-exonic genomic regions into several types based entirely on the exon boundary annotations from General Feature Format (GFF) files http://www.sanger.ac.uk/Projects/C_elegans/WORMBASE/GFF_files.shtml. We defined nine types of regions, based on the exons surrounding the region (Fig. (Fig.3).3). These region types were used only to label the final hits, and were not used in the algorithm to rank them or filter them. They are included merely to aid the user who wants to further inspect the results manually. We excluded exonic regions from our analysis since a) they are unlikely to contain transcription factor bindings sites and b) the high conservation of coding sequences between species would negate the utility of "phylogenetic footprinting".

Classification of non-exonic regions. A hypothetical gene arrangement is shown. "5' intergenic": between exon1 and exon1 of two separate genes; "3' intergenic": between the last exon of both genes. "5'/3' intergenic": between first exon of one gene and last exon of the other gene; "intronic#": between any two exons of one gene; "other": all other possible combinations. In cases where the gene flanking a segment is known to exhibit alternative splicing, the segment was prefixed with 'alt_', i.e. 'alt_intronic#', 'alt_3'intergenic', etc. Two other categories, BEGIN and END, denote regions at the beginning or ending of the chromosome, in the case of C. elegans, or of the sequencing reads in the case of C. briggsae. There were two exceptions to the procedure. The first was due to the fact that the C. briggsae genome we used was an unassembled collection of 578 individual sequence reads. 112 of these reads had no exon annotations, and were ignored in this study. Of these 112, only two were greater than 10,000 bases long, with an average length of 3679.3 nucleotides. Secondly, there were 16 C. elegans and 35 C. briggsae exon annotations one nucleotide long. By visual inspection, we determined that for C. elegans these exons were in fact longer than one nucleotide, but noncoding: in all cases the single nucleotide is 'A' and when spliced forms a TGA stop codon. They were treated as non-existent for this study, which has very little effect on the procedure except that the last true intron of the gene will be considered its 3' region. For C. briggsae, they appear to be errors in the gene annotations and fall within introns. Thus, they were treated as part of the intron in which they occur.
Scanning window scoring procedure
Starting with the input of experimentally defined binding sites of a dimeric homeodomain transcription factor complex composed of the TTX-3 and CEH-10 proteins [16](ASW and OH, submitted), we first use the hidden Markov model software package HMMER [17] with the command 'hmmbuild --null <background-frequency-file> --prior <prior-frequency-file> <output-position-weight-matrix> <input-binding-site-alignment>.' The position weight matrix is an n × 4 matrix where n is the length of the transcription factor binding site alignment input used (in our case, either 14 or 16 nucleotides). The matrix is defined as:
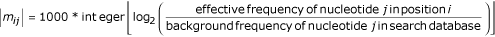
where the effective frequency is determined as the normalized, prior-adjusted, weighted sum of counts of each nucleotide in the alignment columns, in which the sequence weights are determined using a tree-based scheme. We use the resulting position weight matrix as input to CisOrtho. Then, CisOrtho finds approximately the N (option -t) highest scoring hits, with the restriction that no single gene has more than D (option -d) hits. The score for a given sequence window is simply the sum of the n cells of the position weight matrix corresponding to the position and nucleotide of the sequence window. For example, the sequence AACTCG would be given the score m1A + m2A + m3C + m4T + m5C + m6G for a 4 × 6 position weight matrix |mij|. This scoring scheme is simply the log-odds scoring used by the original HMMER software, adapted as a scanning window algorithm via CisOrtho. We note that known target genes of TTX-3 do not contain clustered binding sites [16](ASW and OH, submitted) which allowed us to eschew clustering of binding sites as a search criteria. It is possible that previously described searches for transcription factor targets which used the clustering criteria [2,3] may result in too many false-negative results.
The effect of options -t and -d warrants further comment. To find approximately N (option -t) highest-scoring hits in a large amount of DNA, CisOrtho must first estimate the raw minimum score cutoff which yields this many hits. Technically, this is achieved by scoring every 100th sequence window, finding the top N/100 hits, and using the lowest score of those as the cutoff in the full search. Since the criterion for acceptance is a raw score cutoff, the actual number of hits retrieved in the whole sequence cannot be controlled precisely, but depends on the statistics of the sequence and position weight matrix. Furthermore, since no gene is allowed more than D associated hits (option -d), the total number retrieved will be lower if there is a lot of clustering and thus many genes have more than D actual hits. If the user wants to perform an exhaustive search, the special value of zero for -t indicates using no score cutoff, thus accepting all windows as hits. Likewise, the -d option can be set to an arbitrarily high value (in the downloaded software). These settings may be of interest in preliminary runs to determine the clustering behavior of the hits. More restrictive settings should then be used based on the results of the preliminary run.
Orthology-based filtering and HTML tables
First, CisOrtho retrieves the set of all C. elegans/C. briggsae ortholog pairs provided as a one-to-one mapping in the file orthologs-2.00 ftp://ftp.wormbase.org/pub/wormbase/briggsae/run25_analysis_freeze2.00/orthologues/. Then, for each ortholog pair in this list, CisOrtho finds the highest scoring hit for each of the C. elegans and C. briggsae orthologs, and stores the two hits as a 'hit-pair'. Note that there is no restriction on which region type (3' intergenic, 5' intergenic, etc.) each hit comes from. For example, the C. elegans highest-scoring hit may be 'intronic1' while the C. briggsae hit may be 3'-intergenic. Each hit in the hit-pair is described by the score, nucleotide sequence and type of region in which it occurs. In addition, for each of the directly adjacent genes, the coding strand, gene name and relative position of the hits in relation to the flanking genes are provided (Fig. (Fig.3).3). Finally, for the hit-pair itself, the number of mismatches between C. elegans and C. briggsae hits is given, along with the average, maximum and minimum scores from the pair of hits. Several HTML output files are generated with the information sorted into columns, and each hit-pair appearing as a grouped pair of lines. The HTML tables are each sorted primarily and secondarily by some combination of the mismatch number and average, maximum or minimum score (Fig. (Fig.4).4). The user has also the option to display multiples target sites located in a hit pair (this option takes into account that transcription factors often bind to multiple sites in a promoter)(Fig. promoter)(Fig.22).

Output of the program pipeline. Hits of a search with the TTX-3 consensus binding site is shown. num: number in list. mis: Number of base mismatches between first C. elegans and first C. briggsae hits. segtype: Type of non-exonic region (see Figure Figure3).3). str1/2: negative (N) or positive (P), strand on which the first/second of the two genes that flank the identified target site are located; offset1/2: distance of the target site to the flanking gene(s) (in relation to the start codon if the target site is 5' or located in an intron; in relation to the stop codon if the site is 3' to the gene; in the latter two cases, the number has a positive value); ID: cosmid name of the flanking genes, name: flanking gene names (if available). Gene IDs/names are linked to the WormBase gene model at http://www.wormbase.org, which contains further information about the gene. In case there are multiple target sites located in a defined inter/intragenic region, there is an option to report the n highest scoring hits for each ortholog. If this option is used, the top-scoring C. elegans or C. briggsae hit in each hit-pair will be highlighted, and the next n-1 hits will be gray. Color coding: Orthologous C. elegans/C. briggsae genes ("hit-pairs") are color coded in blue (Y39A3B.5 and CBG15122 are orthologs) and green (M01E10.2 and CBG15118 are orthologs).
Validation of the procedure
TTX-3 is a LIM homeodomain transcription factor that we have previously shown to be required for interneuron differentiation in C. elegans [16]. More recently, we found that TTX-3 binds together with the Paired-type homeodomain transcription factor CEH-10 to a 16 base pair target site, conserved in seven direct TTX-3/CEH-10 target genes (ASW and OH, submitted). We have used CisOrtho to identify new target genes of the TTX-3/CEH-10 homeodomain proteins on a genome-wide level, using as an input the experimentally determined, 16 bp TTX-3/CEH-10 consensus binding site (ASW and OH, submitted). We sorted the list of hit-pairs that CisOrtho provided by average score between the two species. We have experimentally verified hits from this list using either or both of two criteria: When fused to a heterologous reporter gene, the putative target should be expressed in the same neuron type as the ttx-3 gene and the reporter gene should not be expressed in that neuron in ttx-3 null mutant animals [16]. We have generated 15 new reporter gene fusions for the top 26 hits. 14 of these reporter gene fusions satisfy the experimental criteria to represent targets of TTX-3/CEH-10 (ASW and OH, submitted). We have also generated reporter fusions to lower scoring hit-pairs. For example, 11 out of 17 tested predicted sites that ranked between 42 and 112 in the hit-score list were experimentally confirmed to be TTX-3 targets (ASW and OH, submitted).
We have also made use of the C. elegans/C. briggsae mismatch feature of CisOrtho and generated reporter fusions to predicted binding sites with a low score yet almost perfect conservation between C. elegans and C. briggsae. 13/22 tested sites from this "low-mismatch" list fulfilled the experimental criteria to be TTX-3/CEH-10 targets (ASW and OH, submitted). We also note many cases in which conserved binding sites are located in introns (first intron or later introns); in most cases examined by reporter gene fusions, these sites were functional. Finally, we note that the representation of the starting matrix did not significantly change upon inclusion of new, experimentally verified TTX-3/CEH-10 target sites. Taken together, CisOrtho has been successfully used to identify new target genes for a transcription factor.
Conclusions
CisOrtho is complementary to but also extends previous approaches to identify transcription factor targets. Previously described phylogenetic footprinting approaches undertook an unbiased search for phylogenetically conserved sequence patches in non-coding regions, assumed to be transcription factor binding sites [6-8], [11-13]. Providing a complement to these approaches, CisOrtho undertakes a more targeted search, finding phylogenetically conserved regulatory targets of defined transcription factors whose DNA binding site specificity is known. CisOrtho extends previously described approaches to identify target genes for specific transcription factors which did not utilize the phylogenetic footprinting aspect for filtering [2,3], or – in cases where phylogenetic footprinting was used – were not conducted in an unbiased, genome wide manner [10,18]. CisOrtho rather takes advantage of GFF files to allow appropriate annotation and subsequent searching of all genomic non-coding sequence space and produces a user-friendly output from the search. While we have limited our approach to the genomes of C. elegans and C. briggsae, CisOrtho can as easily be applied to other genomes that are annotated in the commonly used GFF format.
Availability
CisOrtho is available through a web interface at http://www.wormbase.org/cisortho (Fig. (Fig.2).2). For users who want to run the program locally, or with a wider range of parameters or different inputs, CisOrtho is freely available as precompiled executables for Windows (win32) and Mac OS X, and as a source code distribution for Unix/Linux with traditional 'configure' script and Makefiles. Also provided in the distribution is the Perl script snip.plx, which initially isolates the non-exonic genomic sequence to be searched by CisOrtho, according to GFF (General Feature Format) genome annotation files. Users experienced in Perl may modify this script to output genomic sequence based on different criteria. For example, if a user wanted to search only 3' regions of genes, this could be implemented by editing snip.plx.
Authors' contributions
HRB wrote the software and web interface, ASW provided the original concept and worked with HRB on improving the program, AW had the original idea to use HMMER software as a scanning window search procedure and wrote an earlier non-ortholog based search program. OH initiated and supervised the study and prepared the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Jack Chen for assistance in installing the website, and Lincoln Stein for generously providing web hosting on WormBase. This work was funded by the NIH.
References
- Bulyk ML. Computational prediction of transcription-factor binding site locations. Genome Biol. 2003;5:201. 10.1186/gb-2003-5-1-201. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Sosinsky A, Bonin CP, Mann RS, Honig B. Target Explorer: An automated tool for the identification of new target genes for a specified set of transcription factors. Nucleic Acids Res. 2003;31:3589–3592. 10.1093/nar/gkg544. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, Rubin GM, Eisen MB. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci U S A. 2002;99:757–762. 10.1073/pnas.231608898. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Tronche F, Ringeisen F, Blumenfeld M, Yaniv M, Pontoglio M. Analysis of the distribution of binding sites for a tissue-specific transcription factor in the vertebrate genome. J Mol Biol. 1997;266:231–245. 10.1006/jmbi.1996.0760. [Abstract] [CrossRef] [Google Scholar]
- Tagle DA, Koop BF, Goodman M, Slightom JL, Hess DL, Jones RT. Embryonic epsilon and gamma globin genes of a prosimian primate (Galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol. 1988;203:439–455. [Abstract] [Google Scholar]
- Cliften PF, Hillier LW, Fulton L, Graves T, Miner T, Gish WR, Waterston RH, Johnston M. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001;11:1175–1186. 10.1101/gr.182901. [Abstract] [CrossRef] [Google Scholar]
- Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE. Human-mouse genome comparisons to locate regulatory sites. Nat Genet. 2000;26:225–228. 10.1038/79965. [Abstract] [CrossRef] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. 10.1038/nature01644. [Abstract] [CrossRef] [Google Scholar]
- GuhaThakurta D, Palomar L, Stormo GD, Tedesco P, Johnson TE, Walker DW, Lithgow G, Kim S, Link CD. Identification of a novel cis-regulatory element involved in the heat shock response in Caenorhabditis elegans using microarray gene expression and computational methods. Genome Res. 2002;12:701–712. 10.1101/gr.228902. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Loots GG, Ovcharenko I, Pachter L, Dubchak I, Rubin EM. rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res. 2002;12:832–839. 10.1101/gr.225502. Article published online before print in April 2002. [Europe PMC free article] [Abstract] [Google Scholar]
- Webb CT, Shabalina SA, Ogurtsov AY, Kondrashov AS. Analysis of similarity within 142 pairs of orthologous intergenic regions of Caenorhabditis elegans and Caenorhabditis briggsae. Nucleic Acids Res. 2002;30:1233–1239. 10.1093/nar/30.5.1233. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- McGuire AM, Hughes JD, Church GM. Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res. 2000;10:744–757. 10.1101/gr.10.6.744. [Abstract] [CrossRef] [Google Scholar]
- Levy S, Hannenhalli S, Workman C. Enrichment of regulatory signals in conserved non-coding genomic sequence. Bioinformatics. 2001;17:871–877. 10.1093/bioinformatics/17.10.871. [Abstract] [CrossRef] [Google Scholar]
- The-C.elegans-Sequencing-Consortium Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–2018. 10.1126/science.282.5396.2012. [Abstract] [CrossRef] [Google Scholar]
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D'Eustachio P, Fitch DH, Fulton LA, Fulton RE, Griffiths-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J, Stajich JE, Wei C, Willey D, Wilson RK, Durbin R, Waterston RH. The Genome Sequence of Caenorhabditis briggsae: A Platform for Comparative Genomics. PLoS Biol. 2003;1:E45. 10.1371/journal.pbio.0000045. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Altun-Gultekin Z, Andachi Y, Tsalik EL, Pilgrim D, Kohara Y, Hobert O. A regulatory cascade of three homeobox genes, ceh-10, ttx-3 and ceh-23, controls cell fate specification of a defined interneuron class in C. elegans. Development. 2001;128:1951–1969. [Abstract] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. 10.1093/bioinformatics/14.9.755. [Abstract] [CrossRef] [Google Scholar]
- Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell. 2003;11:1101–1108. 10.1016/S1097-2765(03)00134-5. [Abstract] [CrossRef] [Google Scholar]
Articles from BMC Bioinformatics are provided here courtesy of BMC
Full text links
Read article at publisher's site: https://doi.org/10.1186/1471-2105-5-27
Read article for free, from open access legal sources, via Unpaywall:
https://bmcbioinformatics.biomedcentral.com/counter/pdf/10.1186/1471-2105-5-27

Citations & impact
Impact metrics
Citations of article over time
Smart citations by scite.ai
Explore citation contexts and check if this article has been
supported or disputed.
https://scite.ai/reports/10.1186/1471-2105-5-27
Article citations
In silico analysis of the transcriptional regulatory logic of neuronal identity specification throughout the C. elegans nervous system.
Elife, 10:e64906, 24 Jun 2021
Cited by: 11 articles | PMID: 34165430 | PMCID: PMC8225391
TargetOrtho: a phylogenetic footprinting tool to identify transcription factor targets.
Genetics, 197(1):61-76, 20 Feb 2014
Cited by: 8 articles | PMID: 24558259 | PMCID: PMC4012501
The microRNA mir-71 inhibits calcium signaling by targeting the TIR-1/Sarm1 adaptor protein to control stochastic L/R neuronal asymmetry in C. elegans.
PLoS Genet, 8(8):e1002864, 02 Aug 2012
Cited by: 42 articles | PMID: 22876200 | PMCID: PMC3410857
The in vivo dissection of direct RFX-target gene promoters in C. elegans reveals a novel cis-regulatory element, the C-box.
Dev Biol, 368(2):415-426, 05 Jun 2012
Cited by: 14 articles | PMID: 22683808
Coordinated regulation of cholinergic motor neuron traits through a conserved terminal selector gene.
Nat Neurosci, 15(2):205-214, 27 Nov 2011
Cited by: 109 articles | PMID: 22119902 | PMCID: PMC3267877
Go to all (26) article citations
Data
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
NemaFootPrinter: a web based software for the identification of conserved non-coding genome sequence regions between C. elegans and C. briggsae.
BMC Bioinformatics, 6 Suppl 4:S22, 01 Dec 2005
Cited by: 1 article | PMID: 16351749 | PMCID: PMC1866385
Evolution of dnmt-2 and mbd-2-like genes in the free-living nematodes Pristionchus pacificus, Caenorhabditis elegans and Caenorhabditis briggsae.
Nucleic Acids Res, 32(21):6388-6396, 02 Dec 2004
Cited by: 32 articles | PMID: 15576683 | PMCID: PMC535690
A novel non-coding DNA family in Caenorhabditis elegans.
Gene, 388(1-2):61-73, 14 Oct 2006
Cited by: 0 articles | PMID: 17134856
The draft genome sequence of the nematode Caenorhabditis briggsae, a companion to C. elegans.
Genome Biol, 4(12):238, 18 Nov 2003
Cited by: 23 articles | PMID: 14659008 | PMCID: PMC329410
Review
 Free full text in Europe PMC
Free full text in Europe PMC


