

Short abstract
A genome-scale metabolic model of E. coli has been reconstructed, including 904 genes and 931 unique biochemical reactions, which gives a more complete and chemically accurate description of E. coli metabolism than previous models.
Abstract
Background
Diverse datasets, including genomic, transcriptomic, proteomic and metabolomic data, are becoming readily available for specific organisms. There is currently a need to integrate these datasets within an in silico modeling framework. Constraint-based models of Escherichia coli K-12 MG1655 have been developed and used to study the bacterium's metabolism and phenotypic behavior. The most comprehensive E. coli model to date (E. coli iJE660a GSM) accounts for 660 genes and includes 627 unique biochemical reactions.
Results
An expanded genome-scale metabolic model of E. coli (iJR904 GSM/GPR) has been reconstructed which includes 904 genes and 931 unique biochemical reactions. The reactions in the expanded model are both elementally and charge balanced. Network gap analysis led to putative assignments for 55 open reading frames (ORFs). Gene to protein to reaction associations (GPR) are now directly included in the model. Comparisons between predictions made by iJR904 and iJE660a models show that they are generally similar but differ under certain circumstances. Analysis of genome-scale proton balancing shows how the flux of protons into and out of the medium is important for maximizing cellular growth.
Conclusions
E. coli iJR904 has improved capabilities over iJE660a. iJR904 is a more complete and chemically accurate description of E. coli metabolism than iJE660a. Perhaps most importantly, iJR904 can be used for analyzing and integrating the diverse datasets. iJR904 will help to outline the genotype-phenotype relationship for E. coli K-12, as it can account for genomic, transcriptomic, proteomic and fluxomic data simultaneously.
Background
Escherichia coli is perhaps the best characterized and studied bacterium and is of interest industrially, genetically and pathologically. For these reasons, in silico modeling efforts have been made to describe and predict its cellular behavior. With the vast amounts of '-omics' data that are being generated, there is a growing need for incorporating and reconciling heterogeneous datasets, including genomic, transcriptomic, proteomic and metabolomic data [1]. A constraint-based model of E. coli metabolism can accomplish this and serve as a model centric database.
In addition to providing the context for various '-omics' data types, constraint-based models provide a framework to compute cellular functions [2]. This modeling method finds the limits of cellular, biochemical and systemic functions, thereby identifying all allowable solutions. Searches within the allowable solution space can identify solutions of interest, for example a solution that maximizes a particular objective. This approach to genome-scale model building has been reviewed in detail [3-6]. In general, the application of successive constraints (stoichiometric, thermodynamic and enzyme capacity constraints), with respect to the metabolic network, restricts the number of possible solutions. Linear optimization is often used to find a particular solution in the allowable solution space that maximizes a chosen objective function, such as cellular growth (Figure 1). A more detailed description of the constraint-based modeling approach can be found in Materials and methods.
Figure 1.
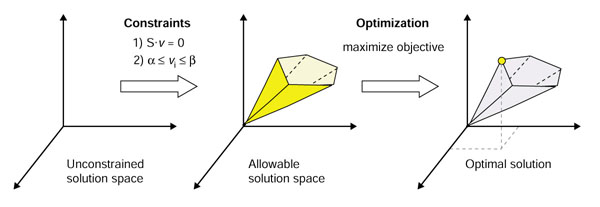
Principles of constraint-based modeling. A three-dimensional flux space for a given metabolic network is depicted here. Without any constraints the fluxes can take on any real value. After application of stoichiometric, thermodynamic and enzyme capacity constraints, the possible solutions are confined to a region in the total flux space, termed the allowable solution space. Any point outside of this space violates one or more of the applied constraints. Linear optimization can then be applied to identify a solution in the allowable solution space that maximizes or minimizes a defined objective, for example ATP or biomass production [3-6].
The constraint-based modeling approach has been used to study E. coli metabolism for over ten years; the history of such model building efforts has recently been reviewed [7]. The first genome-scale metabolic (GSM) model accounting for 660 gene products (iJE660 GSM) was reconstructed using genomic information, biochemical data and physiological data [8]. This genome-scale model has been used to perform in silico gene deletion studies [8] and to predict both optimal growth behavior [9] and the outcome of adaptive evolution [10].
This paper reports an expansion of iJE660a GSM, which itself is a slight modification of the original genome-scale metabolic model (iJE660 GSM) [8]. Gene to protein to reaction (GPR) associations are included directly in the new model (iJR904 GSM/GPR). These associations describe the dependence of reactions on proteins and proteins on genes (Figure 2). The metabolic network described by iJR904 has also changed; individual reactions are now elementally and charge balanced, and a significant number of new genes and novel reactions have been added to the model. iJR904 GSM/GPR accounts for over 904 genes and the 931 unique biochemical reactions the encoded proteins carry out. This paper discusses the effects that these additional reactions have on the predictive capabilities of the model and identifies putative ORFs in the genome which could resolve gaps in the metabolic network.
Figure 2.
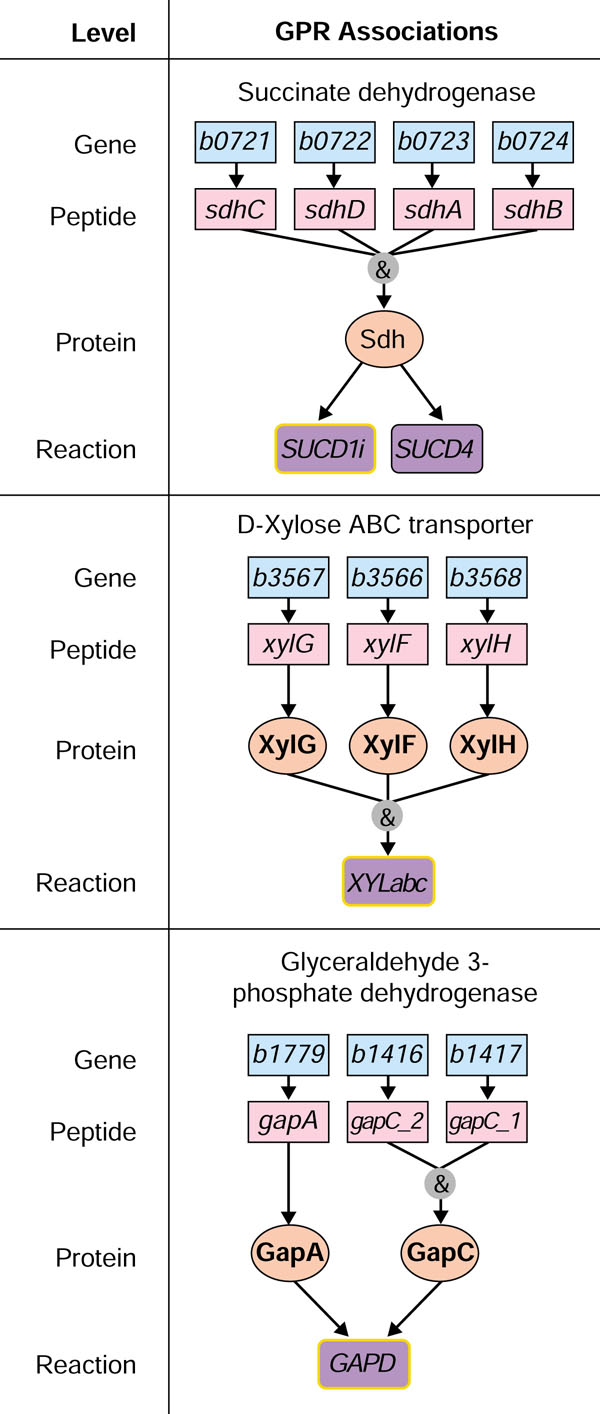
Representation of gene to protein to reaction (GPR) associations. Each gene included in the model is associated with at least one reaction. Examples of different types of associations are shown, where the top layer is the gene locus, the second layer is the translated peptide, the third layer is the functional protein and the bottom layer is the reaction (shown as its corresponding abbreviation listed in the additional data file). Subunits (for example, sdhABCD and gapC_1C_2) and enzyme complexes (for example, xylFGH) are connected to reactions with '&' associations, indicating that all have to be expressed for the reaction to occur. For sdhABCD, the '&' is shown above the functional protein level, denoting that all of these gene products are needed for the functional enzyme. With xylFGH the '&' association is shown above the reaction level, indicating that the different proteins form a complex that carries out the reaction. Isozymes (for example, gapC_1C_2 and gapA) are independent proteins which carry out identical reactions where only one of the isozymes needs to be present for the reaction to occur. Isozymes are shown as two or more arrows leaving different proteins but impinging on the same reaction.
Since computational models of E. coli will continue to grow in size and scope [7] it will become important to be able to distinguish between the different models - a naming convention will aid in this effort. The naming convention we chose to use mirrors the one already established for plasmids. The general form of the names of in silico strains used is iXXxxxa YYY. The 'i' in the name refers to an in silico model (that is, a computer model). This 'i' is followed by the initials (XX) of the person who developed the model and then the number of genes (xxx) included in the model. Any letters (a) after the number of genes indicates that slight modifications were made to the model, for instance iJE660a is derived from iJE660. Further designation of the content and scope of a model are found in YYY; here the acronyms GSM and GPR stand for genome-scale model and gene-protein-reaction associations, respectively. The contents of iJE660a and iJR904 can be found on our website [11], and iJR904 is also detailed in the additional data files.
Results and discussion
Properties of the iJR904 metabolic network
An update on the annotation of the E. coli K-12 genome was published in 2001 [12], facilitating the process of updating the genome-scale in silico E. coli model (iJE660a GSM). Genes encoding known or putative enzymes and transporters not included in the iJE660a model were further examined. Literature and database searches (LIGAND [13], EcoCyc [14] and TC-DB [15]) on each of the genes provided the biochemical information needed to expand E. coli iJE660a. The iJR904 model was built using the software SimPheny™ (Genomatica, San Diego, CA) and accounts for 904 genes with a known locus in the genome, as compared to 660 genes in the previous model.
The metabolic network described by E. coli iJE660a has expanded in size from 627 unique reactions and 438 metabolites to 931 unique reactions and 625 metabolites in iJR904. Complete maps containing all the reactions in the metabolic network are available in the additional data files and can also be downloaded from [11]. The molecular formulae and charges for the metabolites in the model were determined assuming a pH of 7.2. Fifty-eight of the reactions in iJR904 currently do not have associated genes. A complete list of the reactions can be found in the additional data files. Putative functional gene assignments account for 23 of the added reactions, with the majority of these being putative transporters.
In addition to these new reactions, old reactions were updated to be both elementally and charge balanced by including water and protons as participants in the reactions. Six reactions in iJR904 are elementally balanced but not charge balanced (Table 1). Five out of the six reactions are imbalanced because they have not been fully characterized biochemically so assumptions had to be made about the participating metabolites; the remaining reaction (abbreviated ADOCBLS) is charge imbalanced since we were unable to determine the charge associated with the ion complex. The biomass reaction [8], representing the drain of biosynthetic constituents from the network and the growth-associated ATP requirement, was also changed to include internal protons and water. The amount of water needed in the biomass reaction is equal to the amount of ATP hydrolyzed to meet the growth-associated ATP requirement. The hydrolysis of ATP results in the production of a proton while the utilization of NADPH and NADH consumes a proton; this results in the net production of protons in the biomass reaction.
Table 1.
List of charge imbalanced reactions used in iJR904
| Abbreviation | Reaction |
| ADOCBLS | agdpcbi + rdmbzi → adocbl + gmp + h |
| AMPMS | air + h2o → 4ampm + (2) for + (4) h |
| BTS2 | cys-L + dtbt ↔ ala-L + btn + (2) h |
| DHNAOT | dhna + octdp → 2dmmq8 + co2 + h + ppi |
| DKMPPD2 | dkmpp + (3) h2o → 2kmb + for + (6) h + pi |
| MECDPDH | 2mecdp + h → h2mb4p + h2o |
All of these reactions are elementally balanced but not charge balanced (see Additional data file for definition of metabolite abbreviations).
Other updates to the iJE660a reaction network are also notable. A number of reactions in iJE660a could not previously be assigned to an ORF, but are now assigned to an ORF as a result of the updated genome annotation [12], such as mtn and uppS. Other ORF names have changed and these were also updated. Some of the original model reactions were modified in addition to including internal protons and water. These modifications mainly included changes in the stoichiometric coefficients, cofactor usage and reaction reversibility. In some cases, the metabolites that participate in the reaction were changed. Forty-two reactions were removed from iJE660a, and these are listed in the additional data files along with the reasons for their removal.
iJR904 also accounts for the specificity of the quinones in the individual reactions involved in the electron transport chain (Figure 3). E. coli K-12 uses three quinones: ubiquinone (Q), menaquinone (MK) and demethylmenaquinone (DMK) to transfer electrons from the electron donor to the terminal acceptor; in iJE660a there was no distinction between the three quinones and they were all treated as ubiquinone, which led to inaccurate electron donor/electron acceptor pairs.
Figure 3.
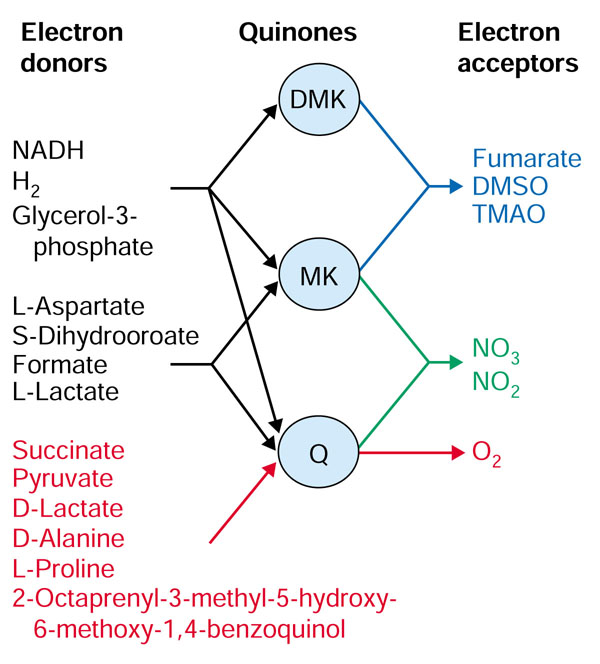
Quinone specificity for electron donors and terminal acceptors. E. coli iJR904 also accounts for the quinone specificity of different reactions. Electron donors are listed on the left, terminal electron acceptors are on the right, and the three types of quinones (DMK, MK, Q; note that these abbreviations differ from those listed in the additional data files) that serve as carriers are shown in the middle. The electron donors shown in red were able to donate their electrons to fumarate in iJE660a, but are now unable to do so. DMK, demethylmenaquinone; DMSO, dimethyl sulfoxide; MK, menaquinone; Q, ubiquinone; TMAO, trimethylamine N-oxide.
GPR associations are for the first time directly included in the iJR904 model, and examples of some are shown in Figure 2. GPR associations have been constructed and their images can be found in the additional data files. These GPR associations can be used to evaluate the reactions remaining in the metabolic network after deletion of a specific gene. Including these associations directly into iJR904 will lead to a more accurate assessment of the effects of gene deletions. In addition, these GPR associations are necessary for analyzing diverse datasets by the model and using these datasets to further identify physiological states.
Thus, iJR904 accounts more accurately for a number of the metabolic processes in E. coli K-12 MG1655, and expands in scope significantly through the addition of GPR associations. E. coli iJR904 GSM/GPR is no longer a purely metabolic model.
Systemic properties
A list of network components alone does not explain how the components work together to produce a biological function. These systemic properties can only be investigated when considering all the components simultaneously; it is the interaction of these components that provides the most information about cellular behavior. As with iJE660a, a myriad of other issues can be addressed with iJR904. We will address three types of issues in this paper using iJR904: gap analysis and putative ORF assignments, the importance of global proton balancing, and phase plane analysis [16].
Identification and resolution of dead ends
A 'dead end' exists in a metabolic network if a metabolite is either only produced or only consumed in the network. If a metabolic network contains a gap, it is missing the biochemical reactions that can produce or consume the dead end metabolites. iJR904 has 70 dead end metabolites or gaps in the network; these are listed in the additional data files. These 70 metabolites participate in 89 reactions, indicating that at least 89 model reactions can never be used if the network is to operate at steady state. The reactions that lead up to the dead ends in iJR904 are included so that when the gaps are filled in at a future date, the network will be fully functional. Some of these network gaps could be reconciled by the addition of transporters, while others could be reconciled by modifying the growth function, that is, including dead end metabolites as requirements for biomass production. Neither of these steps was taken here since genomic or biochemical evidence could not be found to support their inclusion.
Using these gaps, we attempted to identify new functional assignments based on sequence homology searches. A list of EC numbers corresponding to enzymes (not included in the model) that could resolve the gaps, was generated. This list was pooled together with the enzymes known to occur in E. coli which lack assigned loci (EcoCyc [14]). We subsequently collected amino acid sequences from other organisms (orthologous sequences) assigned to these enzymatic functions for a homology search study.
A total of 83 training sets, each with an average of 11 orthologous sequences, was collected and compared against the E. coli genome. Each training set includes multiple orthologous sequences that correspond to an enzyme of interest. Of the 83 training sets, 61 are for enzymes which connect to gaps in the existing network and the remaining 22 are for some of the enzymes listed in EcoCyc. Each training set was processed using the alignment programs MEME [17] and ClustalW [18] to generate a profile for the corresponding enzyme. Using these profiles MAST [19] and HMMER [20] were used to identify similar ORFs in the E. coli genome. Of the 61 enzymes that could resolve network gaps, we assigned putative loci for 12 of them. In addition, putative loci could be found for 15 of the 22 enzymes listed in EcoCyc. Some of these enzymes have multiple matches within the E. coli genome, which together resulted in 55 putative assignments.
These results were inspected manually and found to be relevant and consistent across the three search methods (MAST, with and without end-gap penalty, and HMMER). The results of this study largely coincided with the annotation performed by Serres et al. [12]; however, most annotation updates added more specificity to the type of reactions the enzymes were predicted to catalyze and, in some cases, suggested additional substrates that known enzymes might act upon (Table 2). Table 2 lists the top three matches for each enzyme (except for the case when the match is a known isozyme); a complete list of all results and expected values (e-values) can be found in the additional data files. The putative assignments presented in Table 2 should be used with care. For example, there are multiple putative assignments for the acyl-CoA dehydrogenase enzyme for which the gene has recently been found [21], but the actual gene locus for this enzyme (b0221) does not have the most significant e-value among the list of potential loci.
Table 2.
List of ORFs in E. coli with updated annotations based on sequence similarity
| EC number | Bnum | Gene | Published annotation [12] | Suggested annotation |
| 1.1.1.48 | b1315 | ycjS | Putative NADH-dependent dehydrogenase | D-Galactose 1-dehydrogenase |
| 1.1.1.48 | b1624 | ydgJ | Putative NAD(P)-binding dehydrogenase | D-Galactose 1-dehydrogenase |
| 1.1.1.48 | b3440 | yhhX | Putative NAD(P)-binding dehydrogenase | D-Galactose 1-dehydrogenase |
| 1.1.1.5 | b2426 | ucpA | Putative oxidoreductase, NAD(P)-binding | Acetoin dehydrogenase, diacetyl reductase |
| 1.1.1.5 | b2137 | yohF | Putative oxidoreductase | Acetoin dehydrogenase, diacetyl reductase |
| 1.1.1.5 | b4266 | idnO | 5-Keto-D-gluconate-5-reductase | Acetoin dehydrogenase, diacetyl reductase |
| 1.2.1.19 | b1385 | feaB | Phenylacetaldehyde dehydrogenase | Aminobutyraldehyde dehydrogenase |
| 1.2.1.19 | b1444 | ydcW | Putative aldehyde dehydrogenase | Aminobutyraldehyde dehydrogenase |
| 1.2.1.19 | b3588 | aldB | Aldehyde dehydrogenase B (lactaldehyde dehydrogenase) | Aminobutyraldehyde dehydrogenase |
| 1.2.1.24 | b1385 | feaB | Phenylacetaldehyde dehydrogenase | Succinate-semialdehyde dehydrogenase |
| 1.2.1.24 | b1444 | ydcW | Putative aldehyde dehydrogenase | Succinate-semialdehyde dehydrogenase |
| 1.2.1.24 | b1415 | aldA | Aldehyde dehydrogenase A, NAD-linked | Succinate-semialdehyde dehydrogenase |
| 1.2.7.1 | b1378 | nifJ | Putative pyruvate-flavodoxin oxidoreductase | Pyruvate synthase |
| 1.3.1.2 | b2146 | yeiT | Putative glutamate synthase | Dihydrothymine dehydrogenase |
| 1.3.1.2 | b2147 | yeiA | Putative dihydropyrimidine dehydrogenase, FMN-linked | Dihydrothymine dehydrogenase |
| 1.3.1.2 | b2878 | b2878 | Putative oxidoreductase | Dihydrothymine dehydrogenase |
| 1.3.99.3 | b1695 | ydiO | Putative acyl-CoA dehydrogenase | Acyl-CoA dehydrogenase |
| 1.3.99.3 | b0039 | caiA | Putative acyl-CoA dehydrogenase, carnitine metabolism | Acyl-CoA dehydrogenase |
| 1.3.99.3 | b4187 | aidB | Putative acyl-CoA dehydrogenase; adaptive response (transcription activated by Ada) | Acyl-CoA dehydrogenase |
| 1.6.6.9 | b3551 | bisC | Biotin sulfoxide reductase | Trimethylamine-N-oxide reductase (TMAO reductase II) |
| 1.6.6.9 | b1587 | b1587 | Putative reductase | Trimethylamine-N-oxide reductase (TMAO reductase II) |
| 1.6.6.9 | b1588 | b1588 | Putative reductase | Trimethylamine-N-oxide reductase (TMAO reductase II) |
| 1.6.99.2 | b0046 | yabF | Putative electron transfer flavoprotein-NAD/FAD/quinone oxidoreductase, subunit for KefC K+ efflux system | NAD(P)H dehydrogenase |
| 1.6.99.2 | b3351 | yheR | Putative electron transfer flavoprotein-NAD/FAD/quinone oxidoreductase | NAD(P)H dehydrogenase |
| 1.6.99.2 | b0901 | ycaK | Putative electron transfer flavoprotein-NAD/FAD/quinone oxidoreductase | NAD(P)H dehydrogenase |
| 2.3.3.11 (formerly 4.1.3.9) | b2264 | menD | Bifunctional modular MenD: 2-oxoglutarate decarboxylase and SHCHC synthase | 2-Hydroxyglutarate synthase |
| 2.3.3.11 (formerly 4.1.3.9) | b3671 | ilvB | Acetolactate synthase I, large subunit, valine-sensitive, FAD and thiamine PPi binding | 2-Hydroxyglutarate synthase |
| 2.7.1.23 | b2615 | yfjB | Conserved protein | NAD+ kinase |
| 2.7.1.23 | b3916 | pfkA | 6-phosphofructokinase I | NAD+ kinase |
| 2.7.2.1 | b3115 | tdcD | Propionate kinase/acetate kinase II, anaerobic | Acetate kinase (ackB) |
| 2.7.8.2 | b1408 | b1408 | Putative enzyme | Diacylglycerol cholinephosphotransferase |
| 2.8.1.2 | b2521 | sseA | Putative sulfurtransferase | 3-Mercaptopyruvate sulfurtransferase |
| 2.8.1.2 | b1757 | ynjE | Putative thiosulfate sulfur transferase | 3-Mercaptopyruvate sulfurtransferase |
| 2.8.3.8 | b2222 | atoA | Acetyl-CoA:acetoacetyl-CoA transferase, beta subunit | Acetate CoA-transferase |
| 2.8.3.8 | b2221 | atoD | Acetyl-CoA:acetoacetyl-CoA transferase, alpha subunit | Acetate CoA-transferase |
| 2.8.3.8 | b1694 | ydiF | Putative acetyl-CoA:acetoacetyl-CoA transferase alpha subunit | Acetate CoA-transferase |
| 3.1.1.31 | b0678 | nagB | Glucosamine-6-phosphate deaminase | 6-Phosphogluconolactonase (Pgl) |
| 3.1.1.31 | b3718 | yieK | Putative isomerase | 6-Phosphogluconolactonase (Pgl) |
| 3.1.1.31 | b3141 | agaI | Putative galactosamine-6-phosphate isomerase | 6-Phosphogluconolactonase (Pgl) |
| 3.2.1.68 | b3431 | glgX | Glycosyl hydrolase | Isoamylase |
| 3.5.1.2 | b1524 | yneH | Putative glutaminase | Glutaminase A or B |
| 3.5.1.2 | b0485 | ybaS | Putative glutaminase | Glutaminase A or B |
| 3.6.1.19 | b2954 | yggV | Conserved hypothetical protein | Nucleoside-triphosphate (ITP) diphosphatase |
| 3.6.1.22 | b3996 | yjaD | Conserved hypothetical protein, MutT-like protein | NAD+ diphosphatase |
| 3.6.1.5 | b3615 | yibD | Putative glycosyltransferase | Inosine triphosphate diphosphatase |
| 3.6.1.6 | b3614 | yibQ | Conserved hypothetical protein | Nucleoside (IDP) diphosphatase |
| 4.1.1.39 | b3615 | yibD | Putative glycosyltransferase | Ribulose-bisphosphate carboxylase (2-phosphoglycolate forming) |
| 4.1.2.10 | b0311 | betA | Choline dehydrogenase, a flavoprotein | Hydroxynitrile lyase |
| 4.1.2.10 | b3168 | infB | Protein chain initiation factor IF-2 | Hydroxynitrile lyase |
| 4.2.1.66 | b3022 | ygiU | Conserved protein | Cyanide hydratase |
| 6.2.1.12 | b4069 | acs | Acetyl-CoA synthetase | Hydroxycinnamate-CoA ligase |
| 6.2.1.12 | b1701 | ydiD | Putative CoA-dependent ligase | Hydroxycinnamate-CoA ligase |
| 6.2.1.12 | b2836 | aas | bifunctional multimodular Aas: 2-acylglycerophospho-ethanolamine acyl transferase | Hydroxycinnamate-CoA ligase |
| 6.2.1.4 | b0728 | sucC | Succinyl-CoA synthetase, beta subunit | Succinate-CoA ligase (ITP forming) |
| 6.2.1.4 | b0729 | sucD | Succinyl-CoA synthetase, alpha subunit, NAD(P) binding | Succinate-CoA ligase (ITP forming) |
Annotations shown in bold arose from analyzing the network gaps in iJR904.
Effects of constraining proton exchange flux
The importance of balancing protons internally can now be investigated with iJR904 since the metabolic network accounts for all the protons being generated and consumed by the individual metabolic and transport reactions (only external protons associated with the proton motive force were accounted for in iJE660a). The medium can serve as a pool both supplying and dissipating external protons as needed by the cell. During growth on some carbon sources, the generation of internal protons by the metabolic reactions is relieved by secreting protons into the medium. This subsequently reduces the amount of ATP made by ATPase since these protons could be used to drive this reaction. Under other conditions, a shortage of internal protons is compensated for by taking up protons from the medium and transporting them across the cell membrane into the cytosol.
The effects that the exchange of protons across the system boundary have on predicted growth rates were investigated. A robustness analysis [22], in which the flux through the proton exchange reaction was constrained from its optimal value down to zero, was performed under aerobic conditions for a variety of carbon sources (Figure 4). The predicted growth rates for different carbon sources respond differently as the exchange flux of protons (between the cell and the medium) is reduced to zero; glucose and glycerol were the most sensitive to proton exchange while D- and L-lactate were the least sensitive.
Figure 4.
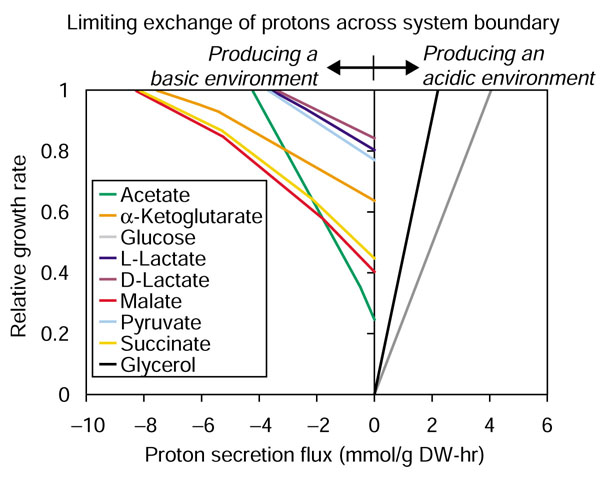
Effect of proton balancing on predicted growth rate. The figure shows how limiting the exchange of protons between the cell and the medium affects the predicted growth rates under aerobic conditions. The relative growth rate (y-axis) is the ratio of the predicted growth rate when the proton exchange flux is limited over the predicted growth rate when the proton exchange flux is not limited (at its optimal value). These calculations are for the conditions of aerobic growth on minimal media; the carbon source uptake rates were set to 5 mmol/g DW-hr and the maximum oxygen uptake rate was set to 20 mmol/g DW-hr. Carbon sources resulting in an outward flux of protons (lines to the right of the y-axis) would make the medium more acidic, while carbon sources resulting in an inward flux of protons (lines to the left of the y-axis) would make the medium more basic.
When either glucose or glycerol was used as the carbon source, excess protons were generated intracellularly; this excess was relieved by secreting extracellular protons, thereby lowering the pH of the medium. For pyruvate, D- and L-lactate, acetate, α-ketoglutarate (αKG), succinate and malate there is a shortage of internal protons; as a result, cells would uptake protons from the medium thereby raising the pH. Since there can be no net accumulation of charge within the system, the total charge entering the system must equal the charge leaving the system. Pyruvate, lactate, acetate, α-ketoglutarate, succinate and malate, as used in these simulations, have a negative charge so H+ must be taken up; however, if the uncharged acidic form of these compounds was used instead (and the acid-base reaction was included in the network - generating proton(s) and the basic form of the compound) it would be predicted that H+ would be secreted by the cell thereby lowering the pH.
It is generally thought that the pH of the medium becomes more acidic as E. coli grows. A lowering of pH has been observed for growth on unbuffered medium with glycerol; however, during growth on unbuffered medium with disodium succinate or sodium acetate the medium becomes more basic (J.L.R. and B.O.P., unpublished data). Clearly iJR904 with charge-balanced reactions highlights the challenge that cells have with globally balancing protons. As a part of the iterative model building process [2]iJR904 can be used to design informative experiments to systematically address this issue.
Phenotypic phase plane (PhPP) comparisons
The phenotypic phase planes [16] for growth on different carbon sources were calculated using both models iJR904 and iJE660a. A description of phenotypic phase planes can be found in Materials and methods. For these simulations the carbon uptake rate and oxygen uptake rate were varied. The carbon substrates tested included: glucose, pyruvate, acetate, glycerol, D-lactate, αKG, succinate and malate. The phase planes for pyruvate, D-lactate and succinate calculated using iJR904 were nearly identical to those calculated with model iJE660a (the lines demarcating the different phases only moved slightly). These results show that the modified and new reactions contained in iJR904 did not significantly affect the phase planes for these substrates, indicating that iJR904 makes similar predictions regarding optimal growth on these substrates.
The line of optimality (LO) on a phenotypic phase plane corresponds to the conditions (oxygen and substrate uptake rates) which can maximize the biomass yield. The largest shifts in the line of optimality were observed for growth on pyruvate and αKG. However, these shifts are relatively small indicating that the optimal oxygen uptake and carbon source uptake rates needed to generate the maximal amount of biomass do not change significantly (less than 10%).
The phenotypic phase planes for carbon sources other than pyruvate, D-lactate, and succinate have more significant changes when calculated with iJR904 as compared to iJE660a. These include growth on the following carbon sources: glycerol, glucose, acetate, malate and αKG. The resulting phase planes calculated using iJR904 and iJE660a are shown in red and blue, respectively, in Figure 5a-e. For the malate and glucose phase planes (Figure 5a,b) one of the lines only appears on the phase plane calculated using iJE660a; these changes are attributed to the effects of global proton balancing described in the previous section. This issue also accounts for changes in the acetate phase plane (Figure 5c) and is a contributing factor to the changes observed with the glycerol and αKG phase planes. However, other changes (discussed below) to the metabolic networks have more significant effects on these two phase planes.
Figure 5.
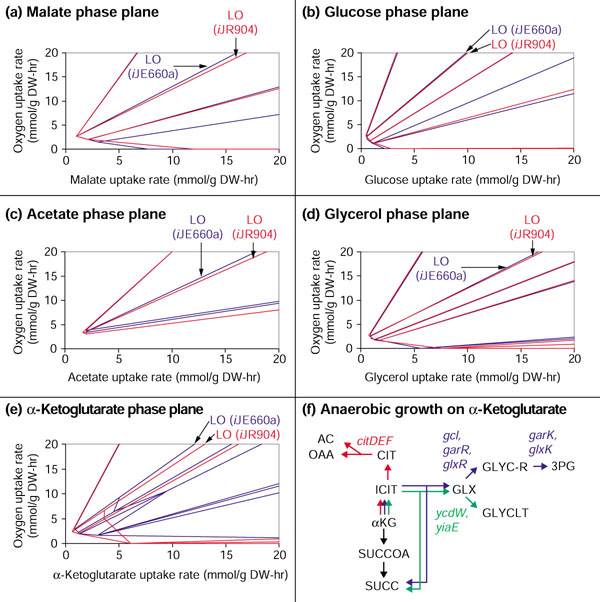
Comparisons between phenotypic phase planes (PhPP) calculated using iJR904 and iJE660a. (a-e) The PhPP for growth on (a) malate, (b) glucose, (c) acetate, (d) glycerol, and (e) α-ketoglutarate (αKG) are shown, where the red lines show the PhPP calculated from iJR904 and the blue lines the PhPP calculated from iJE660a. The line of optimality (LO) corresponds to maximal biomass yield. (e) With αKG as the carbon source, the phase planes calculated from iJR904 (red line) and iJE660a (blue line) are drastically different in the oxygen-limited region (area below the LO). (f) The different metabolic routes (along with their associated genes) added to the network in iJR904 that enable fermentative growth on αKG are shown in red, blue and green. AC, acetate; CIT, citrate; GLYCLT, glycolate; GLYC-R, R-glycerate; GLX, glyoxylate; ICIT, isocitrate; αKG, α-ketoglutarate, OAA, oxaloacetate; 3PG, 3-phospho-D-glycerate; SUCC, succinate; SUCCOA, succinyl-CoA.
Glycerol phase plane
The changes in the glycerol phenotypic phase plane (Figure 5d) were less drastic than those for the αKG phase plane (Figure 5e, see comments below). The only major change was that the lines in the microaerobic region shifted downward. This change is a result of the removal of two reactions involved in pyrridoxal recycling which were previously assigned to pdxH. The two reactions are not included in the metabolic network defined by iJR904 for reasons explained in the additional data files.
α-Ketoglutarate phase plane
The two-dimensional phenotypic phase plane for αKG is noticeably different when calculated for iJR904 and iJE660a (Figure 5e). One notable feature is that iJR904 predicts completely anaerobic growth on αKG, while iJE660a predicts that oxygen is required for growth on αKG. Under oxygen limitations, corresponding to regions below the line of optimality, the expanded model is also more efficient at producing biomass from αKG than the previous model. As oxygen becomes more limiting, iJR904 becomes increasingly more efficient at generating biomass than iJE660a.
Examination of the calculated optimal flux distributions provides insights into why iJR904 is more efficient. One of the reasons for this increased growth efficiency is that iJR904 includes the citrate lyase enzyme, which converts citrate (CIT) to acetate (AC) and oxaloacetate (OAA). During oxygen-limited growth, iJR904 predicts that some of the αKG is converted to OAA by first reversing some of the TCA cycle reactions to generate CIT and then splitting CIT into AC and OAA by citrate lyase. The rest of the αKG is consumed through the forward reactions of the TCA cycle to produce malate (Figure 6). Removal of the citrate lyase reaction from the network under anaerobic conditions shows two other, less-efficient routes that would still enable iJR904 to predict anaerobic growth. These new metabolic routes, which are dependent on at least one of the new additions, are depicted in Figure 5f.
Figure 6.
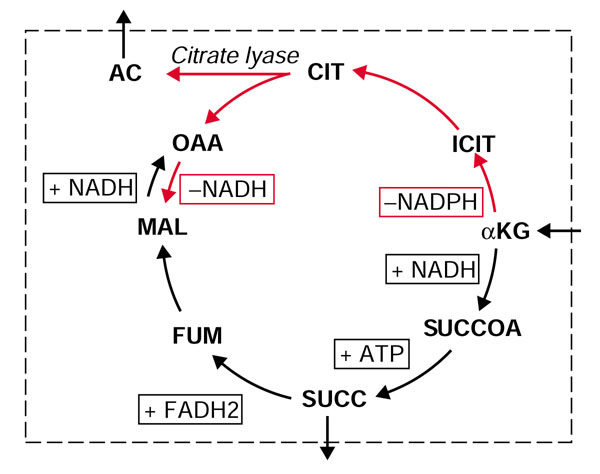
Flux map of TCA cycle and citrate lyase. During oxygen-limited growth on αKG maximal biomass will be made by utilizing the citrate lyase reaction. As oxygen becomes more limiting, the reactions shown in red, culminating in the production of oxaloacetate (OAA), were predicted by iJR904 to be used more heavily than the forward direction of the TCA cycle reactions (shown in black). iJE660a does not include the citrate lyase reaction so carbon flow is directed only in the forward direction of the TCA cycle; these reactions produce more reduced redox carriers, which are difficult for the cell to oxidize in an oxygen-limited environment. The '+' sign indicates that the cofactor is produced by the reaction in the direction shown and the '-' sign indicates that the cofactor is consumed. For abbreviations see Figure 5, with the following additions: ACCOA, acetyl-CoA; FUM, fumarate; MAL, malate, PYR, pyruvate.
Conclusions
This paper reports the curation and expansion of a previous genome-scale constraint-based model of E. coli metabolism (iJE660a GSM) that is now used in multiple laboratories (A.L. Barabasi, personal communication; Church and colleagues [23]; H. Greenberg, personal communication and C. Maranas, personal communication). This expanded model, iJR904 GSM/GPR, includes 37% more metabolic genes and 47% more metabolic reactions. Each reaction in the network is now both elementally and charge balanced with the exception of the six reactions listed in Table 1. While the new reactions added to the network do not change many of the predicted optimal phenotypes, there are instances in which the expanded model makes significantly different predictions, examples of which occur when glycerol, glucose, malate, acetate and αKG are used as the carbon sources under oxygen-limited conditions.
The analysis of dead ends or gaps in the network has led to putative annotations of 55 ORFs. If these putative functional assignments are verified biochemically they can be included in future updates of iJR904. Ideally an iterative process will be developed, within which the model can help identify new targets, and, if verified, can lead to an updated model. This iterative process [2] would be likely to produce more useful results in less-characterized organisms and has already been successful in helping to identify malate dehydrogenase in Helicobacter pylori [24] and citrate synthase in Geobacter sulfurreducens (D. Lovley, unpublished results).
The incorporation of GPR associations into iJR904 will allow for the analysis of transcriptomic and proteomic data directly; it also enables the incorporation of these datasets to further constrain the solution space leading to more accurate predictions of phenotypic data. E. coli iJR904 can now serve as a model centric database which could analyze and reconcile heterogeneous datasets as well as use these datasets to aid in model predictions.
Materials and methods
Constraint-based modeling
A stoichiometric matrix, S (m × n), is constructed where m is the number of metabolites and n the number of reactions. Each column of S specifies the stoichiometry of the metabolites in a given reaction from the metabolic network. Mass balance equations can be written for each metabolite by taking the dot product of a row in S, corresponding to a particular metabolite, and a vector, v, containing the values of the fluxes through all reactions in the network. A system of mass balance equations for all the metabolites can be represented as follows:
![]()
where X is a concentration vector of length m, and v is a flux vector of length n. At steady-state, the time derivatives of metabolite concentrations are zero, and equation (1) can be simplified to:
S • v = 0
It follows that in order for a flux vector v to satisfy this relationship, the rate of production must equal the rate of consumption for each metabolite. Application of additional constraints further reduces the number of allowable flux distributions, v.
Limits on the range of individual flux values can further reduce the number of allowable solutions. These constraints have the form:
α ≤ vi≤ β
where α and β are the lower and upper limits, respectively. Maximum flux values (β) can be estimated based on enzymatic capacity limitations or, for the case of exchange reactions, measured maximal uptake rates can be used. Thermodynamic constraints, regarding the reversibility or irreversibility of a reaction, can be applied by setting the α for the corresponding flux to zero if the reaction is irreversible.
These constraints are not sufficient to shrink the original solution space to a single solution. Instead a number of solutions remain which make up the allowable solution space. Linear optimization can be used to find the solution that maximizes a particular objective function. Some examples of objective functions include the production of ATP, NADH, NADPH or a particular metabolite. An objective function with a combination of the metabolic precursors, energy and redox potential required for the production of biomass has proven useful in predicting in vivo cellular behavior [9,10,25,26].
Simulation conditions
Simulations with iJR904 were all done using the software package SimPheny™ (Genomatica, San Diego, CA); this software was also used to build iJR904. All calculations were made using the conditions outlined in this section. The biomass reaction was the same as that reported previously [8] with the addition of intracellular protons and water, and can be found in the additional data files. All flux values reported in this section are in units of mmol/g DW-hr. The flux through the non-growth associated ATP maintenance reaction (ATPM in the additional data files) was fixed to 7.6. Fluxes through all other internal reactions have an upper limit of 1 × 1030; if the reaction is reversible the lower limit is -1 × 1030 and if it is irreversible the lower limit is zero.
In addition to the metabolic reactions listed in the additional data files, reversible exchange reactions for all external metabolites were also included in the simulations to allow external metabolites to cross the system boundaries. If these exchange reactions are used in the forward direction the external metabolites leave the system and if used in the reverse direction (that is, a negative flux value through the reaction) the external metabolites enter the system.
The following external metabolites were allowed to freely enter and leave the system: ammonia, water, phosphate, sulfate, potassium, sodium, iron (II), carbon dioxide and protons (except during the robustness study where proton exchange flux was constrained down to zero). The corresponding exchange fluxes for these metabolites have a lower and upper flux limit of -1 × 1030 and 1 × 1030, respectively. Aerobic conditions were simulated with a maximum oxygen uptake rate of 20 mmol/g DW-hr, by setting the lower and upper limits for the oxygen exchange flux to -20 and 0 respectively, and anaerobic conditions were simulated by fixing the oxygen uptake rate to 0. All other external metabolites, except for the carbon source, were only allowed to leave the system. The lower and upper limits on their corresponding exchange fluxes were 0 and 1 × 1030, respectively. Growth on different carbon sources was simulated by allowing those external metabolites to enter the system; the actual flux values for uptake rates used in the simulations are noted in the text and figures, where the upper limit is 0 and the lower limit is the negative of the uptake rate listed. These constraints are also summarized in the additional data files.
Phenotypic phase planes (PhPP)
Phenotypic phase plane analysis was developed to generate a global view of the optimality properties of a network [16]. The phenotypic phase plane is constructed from a large number of individual optimal solutions and gives an overall view of the optimality properties of the network. PhPPs are used to show all possible quantitative flux distributions through a network while varying two or three constrained fluxes. The different regions of the phase plane have qualitatively different flux distributions that translate to different metabolic phenotypes. One important feature of the PhPP is the line of optimality (LO). Points that lie on the LO optimize the objective function for a given substrate uptake rate. When the cell is operating along the LO, calculated when the growth rate is used as the objective function, the cell is growing with a maximal biomass yield.
Identification of dead ends
Dead end metabolites are classified as such if a metabolite can either be produced but not consumed or consumed but not produced. By examining the connectivity of the metabolites in the S matrix, a list of dead ends can be generated. Once these are identified, the number of reactions directly involved with these metabolites can be determined by enumerating the number of non-zero elements in the row corresponding to each dead end metabolite.
Sequence annotations
A list of reactions and associated enzymes that could connect the dead ends with the rest of the network was gathered from LIGAND (Database of Chemical Compounds and Reactions in Biological Pathways [13]). These are enzymes known to act on those metabolites in other organisms. The enzymes listed in EcoCyc which are known to be in E. coli but lack assigned loci, were also added to the search list. The enzyme commission numbers for this combined list of enzymes were used in queries against the SwissProt, TremBl and TremBlnew databanks (using Sequence Retrieval System [27]) to retrieve the enzymes' corresponding amino acid sequences. Known orthologous sequences of all of the enzymes of interest were grouped together to construct multiple sequence alignments. Two separate programs, MEME (Multiple Expectation maximization for Motifs Elicitation [17]) and ClustalW [18] were used for this purpose. MEME was run assuming that each sequence may contain a variable number of non-overlapping occurrences of each motif and up to three distinct motifs. Each training set was processed twice with MEME, once with the program's default end-gap penalty and once without it. Default values were used for gap-opening penalty and gap-extension penalty. All sequences in each training set were weighed equally by MEME. ClustalW, however, down-weighed similar sequences in proportion to their degree of relatedness. Pair-wise alignments in ClustalW were run with a dynamic programming algorithm (slow option) and with the Reset Gap option off.
The MEME output files were submitted to MAST [19] (Motif Alignment and Search Tool, version 3.0 online) and searched for matching sequences against the E. coli genome. MAST returned a list of high-scoring sequences and their annotations. ClustalW output files were used by HMMER's hmmbuild and hmmcalibrate (version 2.2g [20]) with default parameters to train profile HMMs (Hidden Markov Models). hmmsearch was subsequently applied to find sequences in the E. coli genome that matched each profile. Results from MAST's and HMMER's searches were manually examined, and relevant matches were reported.
Additional data files
The additional data consists of three files. The Excel file (Additional data file 1) contains the following: a list of the reactions in iJR904; definitions of the metabolite abbreviations; a list of the exchange fluxes used in simulations and their constraints; a list of the dead end metabolites in the metabolic network; a list of the reactions that were not included from iJE660a and a complete list of the sequence comparison results (including e-values). A zip file (Additional data file 2) is also provided, including the JPEG images of all the GPR associations and a detailed document describing how to interpret these images. The GPR image files are each labeled to correspond to either an individual gene or reaction. The third file (Additional data file 3) contains six maps of metabolism which together include all the reactions in iJR904; the reaction and metabolite abbreviations are the same as those listed in the Excel file.
Supplementary Material
A list of the reactions in iJR904; definitions of the metabolite abbreviations; a list of the exchange fluxes used in simulations and their constraints; a list of the dead end metabolites in the metabolic network; a list of the reactions that were not included from iJE660a and a complete list of the sequence comparison results (including e-values)
The JPEG images of all the GPR associations and a detailed document describing how to interpret these images
Six maps of metabolism which together include all the reactions in iJR904
Acknowledgments
Acknowledgements
The authors would like to thank Markus Herrgard for his advice regarding sequence comparisons, Evelyn Travnik for her help putting together the additional data files, as well as Timothy Allen, Jason Papin and Sharon Wiback for their comments on the manuscript. Support for this work was provided by the NIH (grants GM57092 and GM62791) and the San Diego Fellowship to T.V.
References
- Palsson BO. In silico biology through "omics". Nat Biotechnol. 2002;20:649–650. doi: 10.1038/nbt0702-649. [DOI] [PubMed] [Google Scholar]
- Palsson BO. The challenges of in silico biology. Nat Biotechnol. 2000;18:1147–1150. doi: 10.1038/81125. [DOI] [PubMed] [Google Scholar]
- Edwards JS, Covert M, Palsson B. Metabolic modelling of microbes: the flux-balance approach. Environ Microbiol. 2002;4:133–140. doi: 10.1046/j.1462-2920.2002.00282.x. [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BO. Metabolic flux balancing: basic concepts, scientific and practical use. Bio/Technology. 1994;12:994–998. [Google Scholar]
- Bonarius HPJ, Schmid G, Tramper J. Flux analysis of underdetermined metabolic networks: the quest for the missing constraints. Trends Biotechnol. 1997;15:308–314. [Google Scholar]
- Price ND, Papin JA, Schilling CH, Palsson BO. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21:162–169. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- Reed JL, Palsson BO. Thirteen years of building constraint-based in silico models of Escherichia coli. J Bacteriol. 2003;185:2692–2699. doi: 10.1128/JB.185.9.2692-2699.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS, Palsson BO. The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc Natl Acad Sci USA. 2000;97:5528–5533. doi: 10.1073/pnas.97.10.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS, Ibarra RU, Palsson BO. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat Biotechnol. 2001;19:125–130. doi: 10.1038/84379. [DOI] [PubMed] [Google Scholar]
- Ibarra RU, Edwards JS, Palsson BO. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature. 2002;420:186–189. doi: 10.1038/nature01149. [DOI] [PubMed] [Google Scholar]
- Systems Biology Research Group http://systemsbiology.ucsd.edu/organisms/ecoli.html
- Serres MH, Gopal S, Nahum LA, Liang P, Gaasterland T, Riley M. A functional update of the Escherichia coli K-12 genome. Genome Biol. 2001;2:research0035.1–0035.7. doi: 10.1186/gb-2001-2-9-research0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Okuno Y, Hattori M, Nishioka T, Kanehisa M. LIGAND: database of chemical compounds and reactions in biological pathways. Nucleic Acids Res. 2002;30:402–404. doi: 10.1093/nar/30.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Riley M, Saier M, Paulsen IT, Collado-Vides J, Paley SM, Pellegrini-Toole A, Bonavides C, Gama-Castro S. The EcoCyc database. Nucleic Acids Res. 2002;30:56–58. doi: 10.1093/nar/30.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TC-DB: Transport Protein Database http://tcdb.ucsd.edu/tcdb/background.php
- Edwards JS, Ramakrishna R, Palsson BO. Characterizing the metabolic phenotype: a phenotype phase plane analysis. Biotechnol Bioeng. 2002;77:27–36. doi: 10.1002/bit.10047. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- HMMER: Profile HMMs for protein sequence analysis http://hmmer.wustl.edu
- Campbell JW, Cronan JE., Jr The enigmatic Escherichia coli fadE gene is yafH. J Bacteriol. 2002;184:3759–3764. doi: 10.1128/JB.184.13.3759-3764.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS, Palsson BO. Robustness analysis of the Escherichia coli metabolic network. Biotechnol Prog. 2000;16:927–939. doi: 10.1021/bp0000712. [DOI] [PubMed] [Google Scholar]
- Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci USA. 2002;99:15112–15117. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Schilling CH, Famili I, Edwards JS, Goryanin II, Selkov E, Palsson BO. Metabolic modeling of microbial strains in silico. Trends Biochem Sci. 2001;26:179–186. doi: 10.1016/s0968-0004(00)01754-0. [DOI] [PubMed] [Google Scholar]
- Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol. 1994;60:3724–3731. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramanik J, Keasling JD. Stoichiometric model of Escherichia coli metabolism: incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotechnol Bioeng. 1997;56:398–421. doi: 10.1002/(SICI)1097-0290(19971120)56:4<398::AID-BIT6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Zdobnov EM, Lopez R, Apweiler R, Etzold T. The EBI SRS server - recent developments. Bioinformatics. 2002;18:368–373. doi: 10.1093/bioinformatics/18.2.368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of the reactions in iJR904; definitions of the metabolite abbreviations; a list of the exchange fluxes used in simulations and their constraints; a list of the dead end metabolites in the metabolic network; a list of the reactions that were not included from iJE660a and a complete list of the sequence comparison results (including e-values)
The JPEG images of all the GPR associations and a detailed document describing how to interpret these images
Six maps of metabolism which together include all the reactions in iJR904