

Abstract
Human prostate glandular epithelial cells have the unique capability of accumulating high levels of zinc. This is essential to inhibit m-aconitase activity so that citrate can accumulate for secretion into prostatic fluid, which is a major function of the prostate gland. As a result, the Krebs cycle is truncated with the consequence of the lost ATP production that would result from citrate oxidation. The cellular accumulation of zinc also inhibits mitochondrial terminal oxidation and respiration. In addition to these metabolic effects, zinc accumulation exhibits anti-proliferative effects via its induction of mitochondrial apoptogenesis. Zinc accumulation also inhibits the invasive/migration activities in malignant prostate cells. The anti-proliferative effects and the effects on invasion and migration occur through zinc activation of specific intracellular signaling pathways. Consequently, these effects impose anti-tumor actions by zinc. The ability of prostate cells to accumulate zinc is due to the expression and activity of the zinc uptake transporter, ZIP1. To avoid the anti-tumor effects of zinc, in prostate cancer the malignant prostate cells exhibit a silencing of ZIP1 gene expression accompanied by a depletion of cellular zinc. Therefore we regard ZIP1 as a tumor suppressor gene in prostate cancer. In addition to prostate cells, similar tumor suppressor effects of zinc have been identified in several other types of tumors.
Keywords: Zinc, prostate cancer, tumor suppressor, apoptosis, Krebs cycle, zinc transporter, intermediary metabolism
The physiology and biochemistry of zinc and its importance in normal cellular and bodily function has been the subject of numerous reviews [1–4]. The regulation and maintenance of a “normal” concentration and distribution of cellular zinc are essential to the function, metabolism, growth, proliferation and survival of cells. A significant clinical aspect of zinc is its role in the development and progression of malignancy. There is now compelling clinical and experimental evidence that zinc is an important factor in prostate cancer, and also in other types of cancer. In relation to essential tumor cell activities, the effects of zinc can be categorized as: intermediary metabolism and bioenergetics effects; motility and invasive effects; growth and proliferation effects. The actions of zinc on all three activities impose anti-tumor effects in malignant prostate cells and other tumor cells. This review will present the role of zinc in prostate malignancy about which most information exists; and which admittedly has been the focus of the research of the authors. Information has been accumulating that zinc also induces anti-tumor effects in other tumor cells, which will be included in this presentation.
The effects of zinc on the intermediary metabolism of normal and malignant prostate cells
As first reported by the classic studies of Warburg in 1926 [5], virtually all tumor cells exhibit metabolic activities that differ from their precursor sane cells. Although the metabolic alterations might vary in different tumor cells, some common relationships or axioms of cellular activity, cellular metabolism, and malignancy do exist; as follows:
. The existing cellular intermediary metabolism of a cell provides the bioenergetic/synthetic/catabolic requirements that are essential for the manifestation of the cells’ current activities (function, growth, proliferation).
. When the cell’s activity changes, its metabolism must be adjusted consistent with any newly established bioenergetic/synthetic/catabolic requirements.
. Malignant cells are derived from normal cells that have undergone a genetic transformation to a neoplastic cell phenotype that is endowed with malignant potential.
This necessitates a metabolic transformation in the neoplastic cell to provide the bioenergetic/synthetic requirements of malignancy.
. In the absence of the metabolic transformation, the neoplastic cell will not progress to complete malignancy. Conversely, the metabolic transformation, in the absence of the genetic transformation to a neoplastic malignant cell, will not cause malignancy.
Prostate malignancy arises predominantly in the peripheral zone of the prostate gland. The normal secretory epithelial cells are highly specialized cells that synthesize, accumulate and secrete enormously high levels of citrate into the prostatic fluid; which is a major function of the prostate gland. This is achieved by the ability of these cells to accumulate high cellular and mitochondrial levels of zinc. In the mitochondria, the zinc inhibits m-aconitase activity, thereby preventing citrate oxidation via the Krebs cycle (Fig. 1). Thus, the Krebs cycle is truncated prior to the first oxidative step, and citrate is an end-product of intermediary metabolism in the prostate cells; whereas, citrate oxidation is essential in most mammalian cells. The production of citrate as an end-product has bioenergetic implications. The oxidation of glucose up to citrate yields about 14 ATP; from complete glucose oxidation, the yield is about 38 ATP/glucose. Thus loss of citrate oxidation represents about 65% of the potential ATP yield from glucose oxidation that the prostate cell sacrifices for the prostatic function of citrate secretion.
Figure 1.
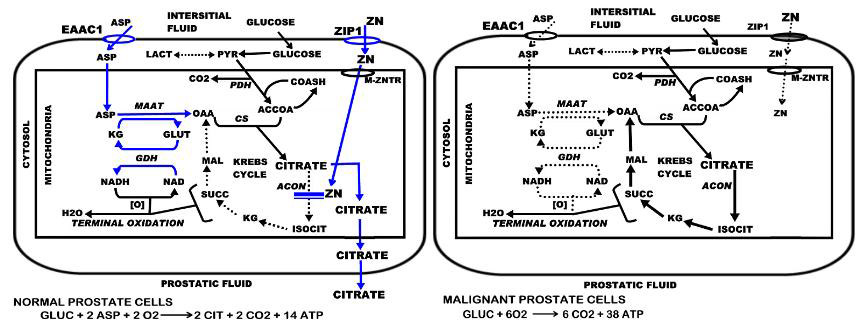
The role of ZIP1 expression and zinc accumulation in the pathway and bioenergetics of citrate production and citrate oxidation in normal versus malignant prostate cells.
In prostate cancer, the malignant cells undergo a metabolic transformation from citrate-producing to citrate-oxidizing cells. This results from the lost ability of the malignant cells to accumulate zinc. The absence of high mitochondrial zinc levels removes the inhibition of m-aconitase activity. Citrate is then oxidized via a functional Krebs cycle, and the typical complete oxidation of glucose restores the efficient ATP production (Fig. 1). For detailed descriptions of the relationships of citrate metabolism and zinc in prostate see recent reviews [6–10].
Zinc transport and metabolic relationships in prostate
Cellular zinc transport in normal and malignant prostate cells
Since zinc is the key to the prostate cell metabolic transformation, the important issue becomes the mechanism of normal epithelial cell accumulation of high zinc levels; and the mechanism for the lost ability of the malignant cells to accumulate high zinc levels. It is now established that the zinc transporter, ZIP1, is responsible for zinc uptake and accumulation in prostate cells [11, 12]. Recent studies using prostate tissue sections have revealed the expression of ZIP1 and high cellular zinc levels in citrate-producing non-malignant prostate glands [13]. In contrast, adjacent malignant glands exhibited down-regulation of ZIP1 gene expression and transporter protein level and depletion of zinc. It is also significant that malignant prostate cell lines derived from metastatic sites (PC-3, LNCaP Du-145) exhibit Zip1 expression and zinc transporter activity [12]. This indicates that ZIP1 down-regulation in the primary site malignant cells in situ is not due to gene deletion or due to a fatal mutation. Thus, the mechanism of ZIP1 gene silencing needs to be elucidated.
Mitochondrial zinc transport
While ZIP1 (and perhaps other zinc uptake transporters) is responsible for the cellular uptake of zinc from circulation, the accumulation of zinc within the mitochondrial is dependent upon other processes. Mitochondria of all cells sequester a significant amount of the cellular zinc, which Dhar [14] estimates to be about 30% in human prostate. Despite this important relationship, little information exists concerning the mechanism of mitochondrial zinc uptake/transport. As zinc is transported to the cytosolic side of the plasma membrane it is immediately bound to cytosolic ligands so that a cytosolic free Zn++ ion pool is essentially non-existent. The cellular concentration of zinc in most cells is about 200 uM; of which free Zn++ exists in pM-fM concentrations, i.e. essentially physiologically non-existent. Therefore the cytosolic source of mitochondrial zinc is derived from mobile reactive ZnLigands that are 10 kDa or less and are capable of permeating the outer mitochondrial membrane pores. These low molecular weight ZnLigands (such as ZnMetallothionein, ZnCitrate, ZnAspartate, ZnHistidine) constitute the source of “mobile reactive zinc” for transport into the mitochondria [14–18]. This pool of zinc is probably in the low uM range. A mitochondrial zinc uptake transporter activity that is capable of importing zinc from these cytosolic ZnLigands has been kinetically identified in prostate and liver mitochondria [17,18]. The putative transporter effectively transports zinc from ZnLigands that have a binding affinity lower than about log Kf=12. For prostate and liver mitochondria, the transporter ZnLigand Km=~30–80 uM depending upon the ligand, which approximates the expected concentration of the zinc donor pool that exists in the cytosol. The mechanism involves the direct intermolecular exchange of zinc from the donor ZnLigand to the recipient putative uptake transporter protein which has an apparent log Kf~11, thus free Zn++ ions are not required for the transport process. The identification of this transporter is a current essential issue to resolve. Whether this putative mitochondrial transporter is down regulated, as is ZIP1, in the malignant cells is not known. However, the decline in the cellular zinc level that occurs in malignant prostate cells would reduce the concentration of the ZnLigands much below the effective Km value; especially since citrate is the major ligand in prostate cells.
Zinc effects on terminal oxidation in prostate cells
Prostate had been characterized as a tissue that possesses a low respiration [19,20]. Consistent with this, the activities of electron transport complexes 1–4 and respiration are markedly lower in prostate mitochondria than those found in other cells [21, 22]. Several reports have identified zinc as an inhibitor of terminal oxidation in mammalian cell mitochondria [23–27]. Because of the uniquely high concentration of zinc in prostate cells, we reported that physiological levels and forms of zinc inhibit the respiration and terminal oxidation of prostate mitochondria [22]. The inhibition occurs at Complex III, and possibly at Complex I and/or II; but no inhibition at Complex IV. The inhibition occurs also with isolated liver mitochondria when supplemented with zinc. Additionally, the constitutive activity levels of complexes I-IV of prostate mitochondria are markedly lower (50–80% lower) than liver mitochondria. The combination of low levels of electron transport components and the inhibitory effect of high zinc accumulation on terminal oxidation are major factors associated with the low respiration of these prostate cells. Consequently, the inhibitory effects of zinc on terminal oxidation implicate a decrease in coupled oxidative phosphorylation production of ATP.
Zinc tumor-suppressor metabolic/bioenergetic effects
The process of high zinc accumulation and its inhibitory effects on citrate oxidation and terminal oxidation are unique metabolic activities that evolved for the citrate producing function of the prostate gland. The normal prostate secretory epithelial cells successfully survive and function under these unique metabolic conditions. For example, the decrease in oxidative production of ATP that accompanies these effects of zinc is compensated for by a high aerobic glycolysis that characterizes these cells. However, these zinc-induced metabolic conditions do not exist in the malignant cells. There exists no substantial evidence of the existence of zinc-accumulating citrate-producing malignant cells in situ in prostate cancer. Essentially all clinical evidence shows that malignant glands consistently exhibit a marked and significant decline of zinc and citrate levels [9, 28]. Moreover the metabolic decrease in zinc and in citrate occurs early in the development of malignancy. Therefore, one must conclude that the metabolic/bioenergetic effects of zinc are incompatible with the ability of the neoplastic cell to conduct its potential malignant activities. To alleviate this imposition, ZIP1 expression is silenced and zinc accumulation is prevented; so as to provide the metabolic/bioenergetic requirements of the malignant process. The recent observation that zinc accumulation and ZIP1 protein expression are lower in the RWPE2 tumorigenic prostate cell line compared to the non-tumorigenic RWPE1 cell line supports the role of zinc and ZIP1 as tumor suppressors [29]. This concept of prostate tumorigenesis is presented in figure 2. Therefore, we propose that ZIP1 is a tumor-suppressor gene, and zinc is an anti-tumor agent in prostate cancer.
Figure 2.
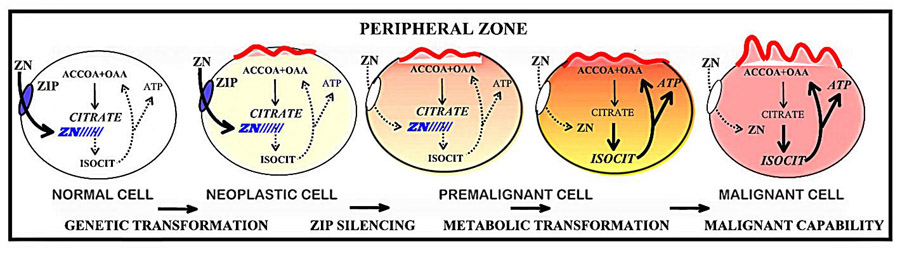
The concept of the genetic and metabolic transformations involved in the development and progression of prostate malignancy; and the importance of ZIP1 expression and cellular zinc accumulation.
THE APOPTOTIC EFFECTS OF ZINC IN PROSTATE CELLS
Tumor cells exist as parasitic cells. As such they have no function other than the generational propagation of their “species”; which is achieved at the expense of their host. Therefore, cell proliferation is an essential activity for the progression of malignancy. A consequence of the cellular accumulation of zinc is an inhibitory effect on proliferation of these prostate cells that results from its induction of apoptosis [30–32]. This effect results from a direct action of zinc on the mitochondria that causes the release of cytochrome c followed by caspase activation and cascading apoptotic events. Our recent studies (unpublished) reveal that zinc exerts a direct effect on mitochondria that facilitates the insertion/docking of Bax and subsequent oligomerization that are associated with the pore-forming process for release of cytochrome c (Fig. 3). Thus, zinc induction of apoptosis in prostate cells results from a mitochondrial apoptogenic effect of zinc.
Figure 3.

Representation of the dual effects of zinc in Bax-associated induction of mitochondrial apoptogenesis in malignant prostate cells. Pathway 1 represents an effect of zinc on Bax expression that increases the cellular level of Bax (TF=transcription factor). Pathway 2 represents the effect of zinc on the mitochondrial insertion and oligomerization of Bax that leads to pore formation for the release of cytochrome c
In addition, zinc causes an increase in the cellular level of Bax, but no corresponding increase in the cellular level of Bcl-2 (fig. 2) [33; unpublished info]. This results in an increase in the Bax/BCL-2 ratio, which is an initiating apoptotic signal in cells. The increase in cellular Bax is attenuated by cyclohexamide and likely due to zinc induction of events that cause increased expression of the Bax gene. The promoter region of the Bax gene is devoid of consensus sequences for a metal-response element (MRE). Therefore a direct interaction of zinc with metal response element binding transcription factor-1 (MTF-1) for activation of an MRE is not an available option for Bax gene regulation by zinc. However, several response elements for transcription factors (TFs) reportedly activated by zinc are present in the Bax promoter. For example, Egr-1 is an immediate-early gene that is induced by zinc [34]. A variant of the hypoxia-inducible factor (HIF-1α) is also induced by zinc [35]. Binding sites for these TFs are present in the Bax promoter. Interestingly, recent evidence has established that artificial TFs containing 3 or 5 Zifs, based on the DNA binding domain of Egr-1/Zif268, selectively regulated Bax gene expression [36]. Thus a mechanism involving zinc induction of TFs with subsequent induction of Bax expression does exist and would be consistent with the time course of zinc-induced Bax expression. This effect of zinc increases Bax availability that enhances the mitochondrial effect of zinc.
It is noteworthy that zinc treatment of mice with developing PC-3 xenograft tumors resulted in inhibition of tumor growth accompanied by increased tumor cell level of Bax and increased apoptosis [unpublished results]. Consequently the accumulation of zinc in prostate cells exhibits anti-proliferation effects. Such effects will prevent the progression of malignancy thereby imposing a tumor-suppressor action against prostate cancer. This is avoided by the decrease in zinc accumulation in the malignant prostate cells.
EFFECTS OF ZINC ON INVASIVE/MIGRATION ACTIVITES OF PROSTATE CELLS
Zinc has also been shown to inhibit the invasive capabilities of malignant prostate cells. Ishii et al. [37] reported that the ability of LNCaP cells to invade Matrigel was strongly suppressed by Zn++. In another study Ishii et al. [38] found that aminopeptidase N purified from human prostate was irreversibly inhibited by low concentrations of zinc; which, they concluded, could be associated with invasive capability. Uzzo et al [39] reported that the suppressive effect of zinc on the angiogenic and metastatic potentials of cancer cells was also mediated through the inhibition of specific pathways that regulate progression of prostate cancer. Therefore, the decrease in cellular zinc levels in the malignant cells is necessary to permit these cells to invade the host tissue and to metastasize to other tissue sites. The inhibition of these activities by zinc imposes another tumor suppressor effect.
TUMOR SUPPRESSOR EFFECTS OF ZINC IN OTHER TUMOR CELLS
Zinc insufficiency has been associated with the potentiation for development of many tumors; and, conversely, zinc treatment inhibits development of the tumors. Fong et al [40, 41] reported that zinc deficiency promotes lingual and esophageal tumorigenesis. Abnet et al [42] determined that high human tissue zinc concentration was strongly associated with a reduced risk of developing esophageal squamous cell carcinoma, which is also consistent with the zinc deficiency potentiation of esophageal tumorigenicity in rodents. Jaiswal and Narayan [43] reported that zinc inhibits the proliferation of colon cancer cells. In head and neck cancers, systemic zinc deficiency was associated with increased tumor size and stage of the cancer [44]. Zinc (and copper) is significantly lower in human hepatoma tissue compared with those in the surrounding "normal" hepatic tissue; and the plasma zinc level is significantly lower in hepatoma subjects compared with the subjects without known liver disease [45]. Studies such as these show a high correlation between zinc availability and the development or progression of specific tumors that are indicative of anti-tumor effects of zinc. It is particularly notable that, as described for malignant prostate cells, zinc treatment exhibits anti-tumor apoptotic effects in other cells. Bae et al reported that zinc accumulation induced apoptosis in choriocarcinoma cells [46] and in human epithelial ovarian cancer cells along with an increase in the cellular level of Bax [47]. Similarly, zinc replenishment in rats rapidly induces apoptosis in esophageal epithelial cells and substantially reduces the development of tumors; and the treatment increases the level of Bax [40]. Rudolf et al [48] reported that zinc acts both directly and indirectly on mitochondria to induce apoptosis in HEP-2 cancer cells. Bae et al [47] also determined the effect of zinc treatment of human epithelial ovarian cancer cells on the m-aconitase activity. They observed that the accumulation of cellular zinc resulted in the inhibition of m-aconitase activity, which confirms this metabolic effect of zinc that we reported for prostate cells.
These reports are indicative of wide-spread anti-tumor effects of zinc among different tumor cells. However, there appears to be a multiplicity of the mechanisms of actions of the zinc effects that are somewhat cell specific. Even the consistent apoptotic/anti-proliferation general effect of zinc appears to occur through a variety of mechanisms, and much more information is required regarding the precise mechanisms that are involved. It is also important to note that reports also exist that show inhibitory effects of zinc on apoptosis in specific cells. Epidemiological studies on the effects of dietary zinc and zinc supplements on the incidence of prostate cancer have reported conflicting results. Studies have reported no effect of zinc [49–51], a protective zinc effect [52] and a moderate increased risk of prostate cancer with long term, high dose zinc supplement use [53]. However, these epidemiological studies did not take into account zinc bioavailability based on diet or potential contaminates of the supplement preparations. The divergent results of epidemiological studies to date indicate that well controlled experimental and clinical studies of the efficacy of zinc alone and in combination with other supplements are required. The reasons and/or mechanisms for the reported divergent effects of zinc also need to be explored. Not withstanding this dilemma, abundant experimental and clinical evidence supports the concept of zinc as a tumor suppressor agent in a variety of cancers in addition to its well-founded anti-tumor effects in prostate cancer.
Zinc and intracellular signaling in cancer
Zinc plays an important role in the proliferation, differentiation and metabolic function of all mammalian cells. It does this through the many zinc binding motifs that are present in the primary structure of proteins that result in structural, catalytic and cocatalytic zinc sites [54]. In addition, zinc should be considered an intracellular signaling molecule similar to calcium. As is true of intracellular calcium, intracellular zinc is homeostaticly maintained at extremely low levels either by sequestration in intracellular vesicles or by binding to intracellular metalloproteins and low molecular weight ligands. Various extracellular signals e.g., redox stress, cytokines and growth factors stimulate the release of zinc from metallothionein or alter the transport of zinc which alters the intracellular level of mobile reactive zinc [55]. Zinc then binds to and activates metal-responsive transcription factors or interacts directly with intracellular signaling molecules to modulate the expression of zinc responsive genes and to regulate specific signal transduction pathways. Zinc has been reported to inhibit Ras signaling [56]. This is a function of zinc that is conserved, since homologous mammalian and non-mammalian zinc export transporters activate Ras by decreasing the intracellular level of zinc. Zinc has also been shown to induce apoptosis by an ERK-dependent pathway that involves specific activation of H-Ras, but not N-Ras or K-Ras in IIC9 fibroblast cells [57]. Mutations that activate H-Ras are oncogenic in most cells and lead to transformation [58–60], thus the observation that zinc induced apoptosis involves Ras activation must be reconciled with its oncogenic action. One possible explanation is that the oncogenic action of Ras involves mitogen activation of Ras through plasma membrane receptors. Ras activation by zinc, on the other hand, appears to occur intracellularly, since the effect is enhanced by pyrithione ionophore [57]. Intracellular Ras activation could result in access to addition components that are not available at the membrane. Clearly, addition studies are required to resolve the mechanism of the differential regulation of growth and apoptosis by zinc through Ras.
The mechanism for zinc regulation of signaling pathways is not well understood; however, recent studies suggest that zinc stimulates the activity of kinases in specific signaling pathways. Physiological levels of zinc in the presence of the zinc ionophore pyrithione increased phosphorylation of Akt at threonine 308 and serine 473 which resulted in activation of Akt [61]. Zinc is reported to inhibit TNF-α induced activation of nuclear factor κB (NFκB) while activating AP1 in PC-3 and Du-145 prostate cancer cells [39]. Zinc also increased the phosphorylation of ERK1/2, p38 and JNK. In immortalized embryonic hippocampal H19-7 cells on the other hand, zinc increased the activity of NFκB [62]. The mechanism of NFκB activation involved zinc induced activation of IκB kinase (IKK) which phosphorylates and inactivates the inhibitor protein IκB which allows activated NFκB to enter the nucleus. The inconsistent and variable effects reported for zinc are dependent on a number of factors, such as the cell type, the concentration of zinc, and other conditions employed in the studies. An important unresolved issue is the identification and characterization of the factors that determine if the response of a specific cell type to physiological levels of zinc will be inhibition or induction of apoptosis or no response. Obviously this is a complex issue. In prostate we propose that the high level of zinc contributes to the balance of cell survival, proliferation and apoptosis through its effects on intermediary metabolism and intracellular signaling pathways. Moreover, the loss of zinc accumulation by the prostate results in the loss of these effects and the development and progression of prostate cancer.
Acknowledgments
The studies of the authors contained in this presentation were supported by NIH Grants DK42839, CA71207, CA79903, CA93443.
Footnotes
Abbreviations used: ERK, extracellular signal-regulated kinase; IκB, inhibitor of kappa light chain enhancer in B cells; JNK, c-jun N-terminal kinase; ZIP1, Znt/Int like protein
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vallee BL. Physiol Rev. 1992;39:443–490. doi: 10.1152/physrev.1959.39.3.443. [DOI] [PubMed] [Google Scholar]
- 2.Vallee BL, Falchuk KH. Physiol Rev. 1993;73:79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- 3.Maret W. Biochemistry. 2004;43:3301–3309. doi: 10.1021/bi036340p. [DOI] [PubMed] [Google Scholar]
- 4.Liuzzi JP, Cousins RJ. Annual Review of Nutrition. 2004;24:151–172. doi: 10.1146/annurev.nutr.24.012003.132402. [DOI] [PubMed] [Google Scholar]
- 5.Warburg O, Wind F, Negelein E. Klin Woch. 1926;5:829–832. [Google Scholar]
- 6.Costello LC, Franklin RB. Mol Cell Biochem. 2005;280:1–8. doi: 10.1007/s11010-005-8841-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franklin RB, Milon B, Feng P, Costello LC. Frontiers in Bioscience. 2005;10:2230–2239. doi: 10.2741/1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costello LC, Feng P, Franklin RB. Mitochondrion. 2005;5:143–153. doi: 10.1016/j.mito.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costello LC, Franklin RB. Mol Cancer. 2006;5:17. doi: 10.1186/1476-4598-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costello LC, Franklin RB. Oncology. 2001;59:269–282. doi: 10.1159/000012183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costello LC, Liu Y, Zou J, Franklin RB. J Biol Chem. 1999;274:17499–504. doi: 10.1074/jbc.274.25.17499. [DOI] [PubMed] [Google Scholar]
- 12.Franklin RB, Ma J, Zou J, Guan Z, Kukoyi BI, Feng P, Costello LC. J Inorg Biochem. 2003;96:435–442. doi: 10.1016/s0162-0134(03)00249-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franklin RB, Feng P, Milon BC, Desouki MM, Singh KK, Kajdacsy-Balla A, Bagasra O, Costello LC. Mol Cancer. 2005;4:32. doi: 10.1186/1476-4598-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhar NK, Goel TC, Dube PC, Chowdhury AR, Kar AB. Exp Mol Pathol. 1973;19:139–142. doi: 10.1016/0014-4800(73)90073-7. [DOI] [PubMed] [Google Scholar]
- 15.Maret W. Proc Natl Acad Sci U S A. 1994;91:237–41. doi: 10.1073/pnas.91.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye B, Maret W, Vallee BL. Proc Natl Acad Sci U S A. 2001;98:2317–22. doi: 10.1073/pnas.041619198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costello LC, Guan Z, Franklin RB, Feng P. J Inorg Biochem. 2004;98:664–666. doi: 10.1016/j.jinorgbio.2004.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guan ZX, Kukoyi B, Feng P, Kennedy MC, Franklin RB, Costello LC. J Inorg Biochem. 2003;97:199–206. doi: 10.1016/s0162-0134(03)00291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huggins C. Harv Lect. 1946;52:148–193. [Google Scholar]
- 20.Muntzing J, Varkarakis MJ, Saroff J, Murphy GP. J Med Primatol. 1975;4:245–251. doi: 10.1159/000459860. [DOI] [PubMed] [Google Scholar]
- 21.Costello LC, Franklin RB, Stacey R. Enzyme. 1976;21:495–506. doi: 10.1159/000458902. [DOI] [PubMed] [Google Scholar]
- 22.Costello LC, Guan Z, Kukoyi B, Feng P, Franklin RB. Mitochondrion. 2004;4:331–338. doi: 10.1016/j.mito.2004.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter FE, Ford L. J Biol Chem. 1955;216:357–369. [PubMed] [Google Scholar]
- 24.Kleiner D, von G. JagowFEBS Lett. 1972;20:229–232. doi: 10.1016/0014-5793(72)80802-0. [DOI] [PubMed] [Google Scholar]
- 25.Lorusso M, Cocco T, Sardanelli AM, Minuto M, Bonomi F, Papa S. Eur J Biochem. 1991;197:555–61. doi: 10.1111/j.1432-1033.1991.tb15944.x. [DOI] [PubMed] [Google Scholar]
- 26.Skulachev VP, Chistyakov VV, Jasaitis AA, Smirnova EG. Biochem Biophys Res Commun. 1967;26:1–6. doi: 10.1016/0006-291x(67)90242-2. [DOI] [PubMed] [Google Scholar]
- 27.Nicholls P, Malviya AN. Biochem. 1968;7:305–310. doi: 10.1021/bi00841a038. [DOI] [PubMed] [Google Scholar]
- 28.Costello LC, Franklin RB, Feng P, Tan M, Bagasra O. Cancer Causes Control. 2005;16:901–915. doi: 10.1007/s10552-005-2367-y. [DOI] [PubMed] [Google Scholar]
- 29.Huang L, Kirschke CP, Zhang Y. Cancer Cell Int. 2006;6:10. doi: 10.1186/1475-2867-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang JY, Liu Y, Zou J, Franklin RB, Costello LC, Feng P. Prostate. 1999;40:200–207. doi: 10.1002/(sici)1097-0045(19990801)40:3<200::aid-pros8>3.0.co;2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng P, Liang J, Li T, Guan Z, Zou J, Franklin RB, Costello LC. Mol Urol. 2000;4:31–35. [PubMed] [Google Scholar]
- 32.Feng P, Li TL, Guan ZX, Franklin RB, Costello LC. Prostate. 2002;52:311–318. doi: 10.1002/pros.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng P, Li TL, Guan ZX, Franklin RB, Costello LC. Ann N Y Acad Sci. 2003;1010:316–320. doi: 10.1196/annals.1299.056. [DOI] [PubMed] [Google Scholar]
- 34.Park JA, Koh JY. Journal of Neurochemistry. 1999;73:450–456. doi: 10.1046/j.1471-4159.1999.0730450.x. [DOI] [PubMed] [Google Scholar]
- 35.Chun YS, Choi E, Yeo EJ, Lee JH, Kim MS, Park JW. Journal of Cell Science. 2001;114:4051–4061. doi: 10.1242/jcs.114.22.4051. [DOI] [PubMed] [Google Scholar]
- 36.Falke D, Fisher M, Ye D, Juliano RL. Nucleic Acids Res. 2003;31:e10. doi: 10.1093/nar/gng010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishii K, Otsuka T, Iguchi K, Usui S, Yamamoto H, Sugimura Y, Yoshikawa K, Hayward SWK. Cancer Lett. 2004;207:79–87. doi: 10.1016/j.canlet.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 38.Ishii K, Usui S, Sugimura Y, Yoshida S, Hioki T, Tatematsu M, Yamamoto H, Hirano K. Int J Cancer. 2001;92:49–54. [PubMed] [Google Scholar]
- 39.Uzzo RG, Crispen PL, Golovine K, Makhov P, Horwitz EM, Kolenko VM. Carcinogenesis. 2006;27:1980–1990. doi: 10.1093/carcin/bgl034. [DOI] [PubMed] [Google Scholar]
- 40.Fong LY, Nguyen VT, Farber JL. J Natl Cancer Inst. 2001;93:1525–33. doi: 10.1093/jnci/93.20.1525. [DOI] [PubMed] [Google Scholar]
- 41.Fong LY, Jiang Y, Farber JL. Carcinogenesis. 2006;27:1489–96. doi: 10.1093/carcin/bgl012. [DOI] [PubMed] [Google Scholar]
- 42.Abnet CC, Lai B, Qiao YL, Vogt S, Luo XM, Taylor PR, Dong ZW, Mark SD, Dawsey SM. J Natl Cancer Inst. 2005;97:301–306. doi: 10.1093/jnci/dji042. [DOI] [PubMed] [Google Scholar]
- 43.Jaiswal AS, Narayan S. J Cell Biochem. 2004;93:345–357. doi: 10.1002/jcb.20156. [DOI] [PubMed] [Google Scholar]
- 44.Prasad AS, Beck FW, Doerr TD, Shamsa FH, Penny HS, Marks SC, Kaplan J, Kucuk O, Mathog RH. J Am Coll Nutr. 1998;17:409–18. doi: 10.1080/07315724.1998.10718787. [DOI] [PubMed] [Google Scholar]
- 45.Liaw KY, Lee PH, Wu FC, Tsai JS, Lin-Shiau SY. Am J Gastroenterol. 1997;92:2260–3. [PubMed] [Google Scholar]
- 46.Bae SN, Kim J, Lee YS, Kim JD, Kim MY, Park LO. Placenta. 2007;28:22–30. doi: 10.1016/j.placenta.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Bae SN, Lee YS, Kim MY, Kim JD, Park LO. Gynecol Oncol. 2006;103:127–36. doi: 10.1016/j.ygyno.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 48.Rudolf E, Rudolf K, Cervinka M. Biofactors. 2005;23:107–20. doi: 10.1002/biof.5520230206. [DOI] [PubMed] [Google Scholar]
- 49.Chang ET, Hedelin M, Adami HO, Gronberg H, Balter KA. J Natl Cancer Inst. 2004;96:1108–1109. doi: 10.1093/jnci/djh206. [DOI] [PubMed] [Google Scholar]
- 50.West DW, Slattery ML, Robison LM, French TK, Mahoney AW. Cancer Causes Control. 1991;2:85–94. doi: 10.1007/BF00053126. [DOI] [PubMed] [Google Scholar]
- 51.Vlajinac HD, Marinkovic JM, Ilic MD, Kocev NI. Eur J Cancer. 1997;33:101–107. doi: 10.1016/s0959-8049(96)00373-5. [DOI] [PubMed] [Google Scholar]
- 52.Kristal AR, Stanford JL, Cohen JH, Wicklund K, Patterson RE. Cancer Epidemiology. Biomarkers & Prevention. 1999;8:887–892. [PubMed] [Google Scholar]
- 53.Leitzmann MF, Stampfer MJ, Wu K, Colditz GA, Willett WC, Giovannucci EL. J Natl Cancer Inst. 2003;95:1004–1007. doi: 10.1093/jnci/95.13.1004. [DOI] [PubMed] [Google Scholar]
- 54.Auld DS. Biometals. 2001;14:271–313. doi: 10.1023/a:1012976615056. [DOI] [PubMed] [Google Scholar]
- 55.Cousins RJ, Liuzzi JP, Lichten LA. J Biol Chem. 2006;281:24085–24089. doi: 10.1074/jbc.R600011200. [DOI] [PubMed] [Google Scholar]
- 56.Bruinsma JJ, Jirakulaporn T, Muslin AJ, Kornfeld K. Dev Cell. 2002;2:567–578. doi: 10.1016/s1534-5807(02)00151-x. [DOI] [PubMed] [Google Scholar]
- 57.Klein C, Creach K, Irintcheva V, Hughes KJ, Blackwell PL, Corbett JA, Baldassare JJ. Apoptosis. 2006 doi: 10.1007/s10495-006-0089-6. [DOI] [PubMed] [Google Scholar]
- 58.Cullen PJ, Lockyer PJ. Nat Rev Mol Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 59.Hancock JF. Nat Rev Mol Cell Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 60.Malumbres M, Barbacid M. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 61.Min YK, Lee JE, Chung KC. Exp Cell Res. 2007;313:312–321. doi: 10.1016/j.yexcr.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 62.Min YK, Park JH, Chong SA, Kim YS, Ahn YS, Seo JT, Bae YS, Chung KC. J Neuroscience Res. 2003;71:689–700. doi: 10.1002/jnr.10520. [DOI] [PubMed] [Google Scholar]