

Abstract
The cyanobacterium, Synechocystis sp. PCC 6803, was the first photosynthetic organism whose genome sequence was determined in 1996 (Kazusa strain). It thus plays an important role in basic research on the mechanism, evolution, and molecular genetics of the photosynthetic machinery. There are many substrains or laboratory strains derived from the original Berkeley strain including glucose-tolerant (GT) strains. To establish reliable genomic sequence data of this cyanobacterium, we performed resequencing of the genomes of three substrains (GT-I, PCC-P, and PCC-N) and compared the data obtained with those of the original Kazusa strain stored in the public database. We found that each substrain has sequence differences some of which are likely to reflect specific mutations that may contribute to its altered phenotype. Our resequence data of the PCC substrains along with the proposed corrections/refinements of the sequence data for the Kazusa strain and its derivatives are expected to contribute to investigations of the evolutionary events in the photosynthetic and related systems that have occurred in Synechocystis as well as in other cyanobacteria.
Keywords: genome sequence; massive parallel sequencer, substrain, Synechocystis sp. PCC 6803
1. Introduction
Cyanobacteria are capable of oxygenic photosynthesis; they are thought to be the progenitor of plant plastids. Synechocystis sp. PCC 6803 is one of the most widely used cyanobacterial species for genetic studies for several major reasons; (i) it is naturally competent by incorporating exogenous DNA into cells that is integrated into the genome by homologous recombination at high frequency;1–3 (ii) it grows heterotrophically in the presence of glucose;3,4 (iii) the entire genome sequence was determined early on by Kaneko et al.5 The availability of the entire genome sequence facilitated post-genomic investigations such as transcriptome-, proteome-, and functional genomics studies.6
The original strain of Synechocystis was isolated from California freshwater by Kunisawa and colleagues and called the Berkeley strain;7 it was deposited in the Pasteur Culture Collection (PCC strain) and the American Type Culture Collection (ATCC strain). Williams3 subsequently isolated the glucose-tolerant (GT) strain from the ATCC strain.3 The Kazusa strain, whose genome sequence was published in 1996,5 is a derivative of a GT strain. A single representative clone of the GT strain was established for complete genome sequencing as the Kazusa strain; other strains were maintained and transferred without further cloning such as single colony isolation.8 Therefore, four substrains, PCC-, ATCC-, GT-, and Kazusa strains, all derived from the original Berkeley strain, were distributed to a number of laboratories although all of them were grouped together under the name Synechocystis sp. PCC 6803.9 Ikeuchi and Tabata8 reported that each substrain had specific mutations such as single nucleotide polymorphisms (SNPs) and indels, and some exhibited a specific phenotype. Some of the mutated loci that were different from the sequence in the database derived from the Kazusa strain have been identified such as a SNP,10 indels,6,11–13 and IS mobilization.14 However, the total number of mutations in the whole genome of each strain remained unknown and sequence variations in these major strains can be expected to raise problems in the evaluation of phenotypes of mutants constructed from these strains. The history of these major substrains and additional substrains, isolated as a single colony from the PCC- and GT strain (GT-I strain; the standard strain in Dr Ikeuchi's group) was summarized by Ikeuchi and Tabata.8 The single colonies isolated from the PCC strain were designated PCC-P (positive phototaxis) strain and PCC-N (negative phototaxis) strain based on the direction of phototactic movement.15 A derivative of the GT-I strain that acquired high light tolerance and a glucose-sensitive phenotype was designated the WL strain, which has an SNP in the pmgA gene.16,17 Thus, there are two fundamental problems for post-genomic research in bacterial molecular genetics. One is the heterogeneity of cells in the frozen stock of the culture collection centres; the other is the frequent spontaneous mutation in bacterial genomes, an event that may be unavoidable during the long cultivation of bacterial cells. As revealed in Bacillus subtilis, whole-genome resequencing is a powerful solution for obtaining the sequence information of such spontaneous mutants.18
Without question, laboratories should start their post-genomic research with genome sequence data of the ‘reference’ or ‘standard’ strain. We deciphered the three substrains of Synechocystis sp. PCC 6803, i.e. PCC-P, PCC-N, and GT-I, to reconstruct the informatics basis of the molecular biology of Synechocystis sp. PCC 6803. We identified a number of SNPs and indels in these substrains and introduced a genetic strategy to identify the mutated loci on a genome-wide level using the massive parallel sequencer. Especially, determination of the genome sequence of PCC substrains will widely contribute cyanobacterial researches using the frozen stock cells supplied from the PCC.
2. Materials and Methods
2.1. Bacterial strains and genomic DNA
Synechocystis PCC-P, PCC-N, and GT-I strains were maintained as frozen stocks in the laboratory of Dr Masahiko Ikeuchi at The University of Tokyo, Japan. The PCC-P and PCC-N strains that exhibited positive- or negative-direction movement under phototaxis test conditions, respectively, were isolated by Yoshihara et al.19 as a single colony from frozen stock obtained from the French Pasteur Culture Collection (PCC strain; see catalogue of strains.9). Genomic DNA was extracted with the hot-phenol method.16
2.2. Sequencing methods
DNA was uniformly sheared into 300-bp portions using Adaptive Focused Acoustics (Covaris Inc., Woburn, MA, USA). We constructed a DNA library with a median insert size of 300 bp for a paired-end read format. The quality of the DNA library was checked with the Sanger method by Escherichia coli transformation of aliquots of the library solution. The library was sequenced on a Genome Analyser II (Illumina Inc., San Diego, CA, USA). Sample preparation, cluster generation, and 50-base paired-end sequencing were according to the manufacturer's protocols with minor modifications (Illumina paired-end cluster generation kit GAII ver. 2, 36-cycle sequencing kit ver. 3) with multiplex method using the single lane of the 8 lane flow-cell. Image analysis and ELAND alignment were with Illumina's Pipeline Analysis software ver. 1.6. Sequences passing standard Illumina GA pipeline filters were retained.
2.3. Mapping analyses using short-read sequences
For short-read alignment and calling variants (SNPs and InDels), we used the short-read mapping software MAQ version 0.7.1,20 BWA ver. 0.5.1,21 and SAMtools ver. 0.1.9.22 MAQ alignments were done using the ‘easyrun’ option of the maq-pl script using the default parameter settings. The SNP filtering was performed using the default parameters except for the minimum consensus quality for SNPs (-q 40). BWA alignments and the subsequent variants calling using SAMtools were done using the default parameter settings. Finally, we applied the following filtering criteria to the lists of SNPs/indels: minimum read depth for SNPs calling = 3, minimum read depth for indel calling = 10, and a 60% cut-off of the percent of aligned reads calling the SNP/indel per total mapped reads at the non-reference allele sites. We also used BWA to estimate the sequence read depth affecting the coverage and accuracy of the variant calls. Structural variations were identified using BreakDancer23 with default parameters.
2.4. Mapping analyses using contigs assembled de novo
Read sequences were assembled de novo with the Velvet assembly programme.24 For optimization of the hash value of the assembly process we used the N50 size. The de novo assembled contigs were mapped on the genome sequence of a database derived from the Kazusa strain using MUMmer sequence alignment package25 with default settings. We then employed show-SNPs functions of the MUMmer program to produce lists of SNPs/indels which were applied the filtering criteria describe above.
2.5. Annotation and creation of the SNP/indel list
The list of SNPs/indels was then annotated with in-house developed software Variant Annotator (VA) that was specifically designed to check amino acid substitutions attributable to the large number of identified SNPs/indels using Genbank annotation files. We used the GenBank, RefSeq, cyanobacterial database Cyanobase (http://genome.kazusa.or.jp/cyanobase), CyanoClust,26 and also ORF information of the GT-S strain,27 which is the recently resequenced substrain of Synechocystis sp. PCC 6803, for precise annotation of each ORF.
2.6. Capillary sequencing with the Sanger method for SNP/indel confirmation
About 200-base genomic regions around the SNPs and indels called by the mapping programmes were amplified by PCR and sequenced on a capillary sequencer with the Sanger method using the commercial sequence service of MACROGEN (Tokyo, Japan). To confirm the SNPs located near IS elements or repetitive regions, the longer DNA fragments were amplified to avoid the amplification of other homologous regions in the Synechocystis genome. The primers used for confirmation are listed in Supplementary Table S1.
2.7. Uploading the genome sequence in the database
Short-read data, obtained on a Genome Analyzer II (Illumina Inc., San Diego, CA, USA), of the substrains of Synechocystis sp. PCC 6803, PCC-P, PCC-N, and GT-I, were deposited in the DRA (DDBJ Sequence Read Archive; http://trace.ddbj.nig.ac.jp/DRASearch/); the accession number is DRA000401. The genome sequences and gene annotations of the substrain GT-I, PCC-P, and PCC-N were also deposited in the DDBJ/GenBank/EMBL database with the accession numbers for each substrain, GT-I (AP012276), PCC-P (AP012278), and PCC-N (AP012277).
2.8. Phylogenetic analysis of Synechocystis sp. PCC 6803 substrains
Phylogenetic relationship of various strains was estimated by the maximum parsimony method by assuming that both base change and indel are treated as a single event. The computation was performed by the dolpenny software of the Phylip package version 3.67,28 using the polymorphism option. Each branch length was set as the number of events occurring along the branch.
3. Results
3.1. Analytical scheme applied to the massive short-read data obtained by next-generation sequencing
The amplified DNA library was sequenced by GAII with 50-base paired-end methods using the parameter settings described in Materials and methods section. We obtained 250, 257, and 221 Mb read data for GT-I, PCC-N, and PCC-P substrains, respectively (Table 1). These read depths correspond to more than 60 times the genome size of Synechocystis. Read data were mapped using three analyses to identify the genomic position of the SNPs, indels, and rearrangements (Fig. 1): (i) BWA21 and MAQ20 for mapping analysis using raw read data; (ii) Velvet24 and MUMmer25 for mapping analysis using de novo assembled contigs; and (iii) BreakDancer23 for rearrangement analysis such as IS movement. The number of mutations was called by each programme and passed through the filter settings of the SAMtools program22 (Table 1). Distributions of averaged read depth in each 1 kb along with the entire genome, obtained by BWA and MAQ were shown in Supplemental Fig. S1. At least 15 times read depth was obtained even in the lowest read depth region. It also indicates that patterns of the read depth distribution depend on the algorithm of each mapping programme. The mapping programmes BWA, MAQ, and MUMer called 76, 69, and 85 potential mutation points, respectively, for the GT-I strain as primary data, 89, 75, and 109 points for the PCC-P strain, and 78, 79, and 104 points for the PCC-N strain. To confirm these results, we checked the sequence around all these loci by Sanger sequencing; regions of ∼200 bp were amplified around the position of the mutations. Depending on the parameter settings, read depth and sequence specificity, it happens that common SNPs in all three substrains were detected only in one or two substrains in each programme. Even if the SNP is called only in one strain, we performed Sanger sequencing of the same locus in all three strains. Whole oligo DNA primers prepared for PCR reactions are listed in Supplemental Table S1.
Table 1.
Summary of mapping analyses using the read data (BWA, MAQ) or the de novo assembled contigs (Velvet and MUMmer)
|
Synechocystis sp. PCC 6803 substrains |
|||
|---|---|---|---|
| GT-I | PCC-N | PCC-P | |
| Total read bases (Mb) | 250 | 257 | 221 |
| Averaged read depth | 70 | 72 | 62 |
| Genome coverage (%) | 99.99 | 99.99 | 99.99 |
| Mapping programmes | Number of SNPs and indels called by each programmes (Final number of differences/number of differences including false-positive data) | ||
| MAQ | 16/76 | 26/78 | 23/89 |
| BWA | 19/69 | 32/79 | 28/75 |
| Velvet and MUMmer | 22/85 | 33/104 | 29/109 |
| BreakDancer | 3/3 | 3/3 | 3/3 |
| Final number of differences to the database | 28 | 44 | 39 |
Figure 1.

Analytical scheme of the read data obtained by massive parallel sequencing. The preparation of the DNA library is described in Materials and methods section. The mapping programmes BWA and MAQ were used for short-read data; the de novo assembly programme was Velvet, and MUMmer was the mapping programme for assembled contigs.
3.2. Combinatorial use of the mapping programmes contributes to the identification of SNPs and indels
Confirmation by Sanger sequencing alerted to a number of false-positive SNP- and indel-calls. Final numbers of SNPs/indels in each substrain were only about 20–40% of the called numbers by each programme as shown in Table 1. Most of the false-positive SNPs or indels were located in repetitive regions or in highly homologous genes in the Synechocystis genome. We expected that the cut-off value of mapping programmes as 60% is enough to detect a number of heterogeneous SNPs of Synechocystis which has multi-copy genome, because 80% SNPs were called as heterogeneous SNPs by BWA in case of GT-I strain. However, we could not detect any heterogeneous SNPs among them by the Sanger method. It indicates that there are technical problems in expecting the total number of the heterogeneous SNPs in the whole genome using cut-off value settings or base-call percentage data obtained by these mapping programmes. On the other hand, all 16 SNPs called homogeneous SNPs by BWA in GT-I strain were also confirmed by the Sanger method. The total number of mutations confirmed by the Sanger method is shown in Fig. 2 with the number of mutations including false-positive data in parenthesis. These numbers are the sum of results obtained for the three substrains. We found that the combinatorial use of several programmes is necessary for the comprehensive detection of SNPs/indels and for the identification of mutations. Mutation loci detected commonly by all three programmes were more reliable than that detected by only the single programme (Fig. 2). Difference of the distribution pattern of the read depth in each mapping programme also suggests that combination use of several programmes is more adequate (Supplemental Fig. S1). However, it is worth noting, SNPs/indels identified commonly by three programmes covered only 60% of the total number of mutations. The correct detection of IS movements reported by Okamoto et al.14 was possible only with BreakDancer, a programme developed for the detection of genome rearrangements.23
Figure 2.
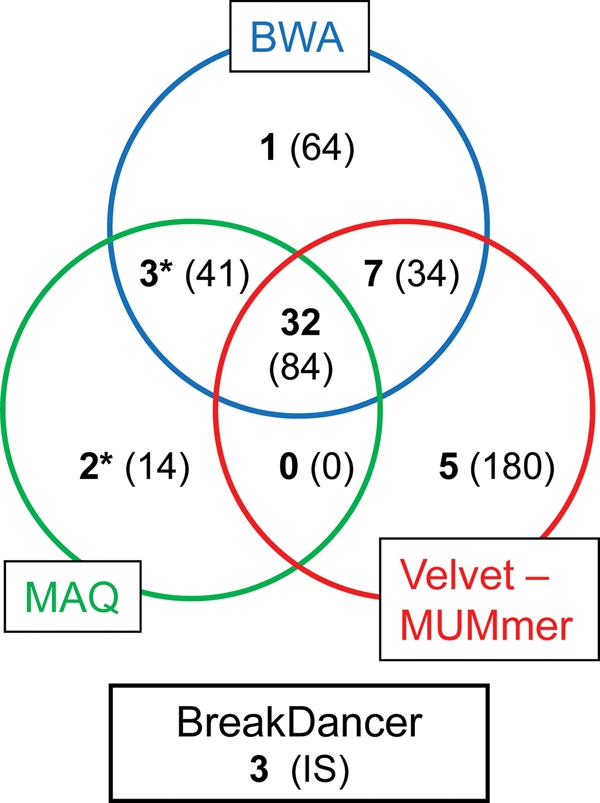
Diagram of the mutations identified by each programme. The number of mutations (SNPs and indels) confirmed by the Sanger method is shown in each circle with the number of mutations including false-positive data in parenthesis. The number of mutations detected by plural programmes is indicated in the circle overlap region. Threshold (cut-off) value of 60% was used in mapping programmes; BWA and MAQ (see Materials and methods section). Mutations detected commonly by all three programmes were more reliable. The combinatorial use of the mapping programmes is important for the genome-wide identification of the mutation loci. Numbers labelled with an asterisk contain miss-called results indicated by parenthesis in Tables 2 and 3.
3.3. Comparison of the identified SNPs/indels and the genome sequence in the database
The confirmed mutations are listed in Tables 2 and 3. The three substrains, GT-I, PCC-P, and PCC-N, manifested at least 22 common different sites compared with the sequence of the Kazusa strain in the database. Among these mutation sites, 15 sites were different from the database sequence, but not real differences in the genomic loci as revealed by Tajima et al.27 (Table 2). We also found that there are totally 14 mutations between the GT/Kazusa- and the PCC-P/PCC-N strains (Table 3). The PCC-P and PCC-N substrains contained three and eight additional specific mutations, respectively (Table 3). These may be potential mutations that elicited the known difference in the phototactic phenotype of the PCC substrains. For example, the PCC-N substrain has mutation in the gspE2 (pilB2) gene for pilus assembly, which also moderately affects the transformation efficiency.15 The PCC-N strain also has a 12-base deletion in the kinase domain of the hik33 gene for the histidine kinase without a frameshift. Hik33 is the multi-stress sensor in Synechocystis and it is conserved in all cyanobacterial species.29–31 This suggests that this substrain may lose the Hik33-dependent regulation of global gene expression, although the relationship between hik33 and phototaxis remains to be determined.
Table 2.
List of the genomic loci of SNPs and indels found in all GT-I, PCC-P, and PCC-N strains compared with the nucleotide sequence in the database
| Genomic loci | Type | Data base | GT-Kazusa | GT-S strain | GT-I strain | PCC-P strain | PCC-N strain | Quality score | Source | Gene ID | Annotation | Amino acid change | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 943495 | SNP | G | A | A | A | A | A | 255 255 | MAQ BWA mummer | slr1834 | psaA | V604I | Smart and McIntosh (10). Error of the Database (27) |
| 1012958 | SNP | G | T | T | T | T | T | 255 255 | MAQ BWA mummer | intergenic region ssl3177-sll1633 | repA-ftsZ | — | Error of the Database (27) |
| 1200143–1201488 (1200306) | Indel (SNP) | IS (C) | IS | — | —(A) | —(A) | —(A) | (108 233) 99 | (MAQ BWA) BreakDancer | sll1780 | ISY203b | — | Insertion of transposase (14). GT-Kazusa specific (27). MAQ and BWA detected this indel region as SNP |
| 1364187 | SNP | A | G | G | G | G | G | 255 255 | MAQ BWA mummer | sll0838 | pyrF | Silent | Error of the Database (27) |
| 2092571 | SNP | A | T | T | T | T | T | 255 255 | MAQ BWA mummer | sll0422 | sll0422 | L313* Stop codon | Error of the Database (27) |
| 2198893 | SNP | T | C | C | C | C | C | 255 255 | MAQ BWA mummer | sll0142 | sll0142 | 689 Silent | Error of the Database (27) |
| 2204584 | Indel | G | G | — | — | — | — | mummer | slr0162 | gspF | gspF+pilC | G-insertion in GT-Kazusa strain causes split of the original pilC gene (34). GT-Kazusa specific (27) | |
| 2301721 | SNP | A | G | G | G | G | G | 255 255 | MAQ BWA mummer | slr0168 | slr0168 | K403E | Error of the Database (27) |
| 2350285–2350286 | Indel | — | A | A | A | A | A | 317 | BWA mummer | intergenic region sml0001-slr0363 | psbI-slr0363 | — | Error of the Database (27) |
| 2360245–2360246 | Indel | — | C | C | C | C | C | 323 | BWA mummer | slr0364 | slr0364 | Frameshift | Error of the Database (27) |
| 2409244 | Indel | C | — | — | — | — | — | mummer | sll0762 | sll0762 | Frameshift | Error of the Database (27) | |
| 2419399 | Indel | T | — | — | — | — | — | 302 | BWA mummer | sll0752 | sll0752 | Frameshift | Error of the Database (27) |
| 2544044–2544045 | Indel | — | C | C | C | C | C | 180 | BWA mummer | ssl0787 | ssl0787 | Frameshift | Error of the Database (27) |
| 2602717 | SNP | C | A | A | A | A | A | 255 255 | MAQ BWA mummer | slr0468 | slr0468 | H82Q | Error of the Database (27) |
| 2602734 | SNP | T | A | A | A | A | A | 255 255 | MAQ BWA mummer | slr0468 | slr0468 | I88N | Error of the Database (27) |
| 2748897 | SNP | C | T | T | T | T | T | 255 255 | MAQ BWA mummer | intergenic region slr0210-ssr0332 | slr0210-ssr0332 | — | Error of the Database (27) |
| 3142651 | SNP | A | G | G | G | G | G | 255 255 | MAQ BWA mummer | sll0045 | sps | 75 Silent | Error of the Database (27) |
| 3260096 | Indel | C | C | — | — | — | — | Mummer | intergenic region sll0529-sll0528 | sll0529-sll0528 | — | GT-Kazusa specific (27) | |
| 3400322–3401506 | Indel | IS | IS | — | — | — | — | 99 | BreakDancer | sll1474 | ISY203g | sll1473+sll1475 | Insertion of transposase. IS-insertion causes split of the original hik32 gene (14). GT-Kazusa specific (27) |
| Genomic loci | Type | Data base | GT-Kazusa | GT-S strain | GT-I strain | PCC-P strain | PCC-N strain | Quality score | Source | Gene ID | Annotation | Amino acid change | Comment |
| 386410- 386411 (386406) | Indel (SNP) | —(T) | — | — | 102 bp (A) | 102 bp (A) | 102 bp (A) | (68) | (MAQ) | slr1084 | slr1084 | 34 amino acids deletion (V77D) | This indel region was called as SNP by MAQ as shown in parentheses. This indel region was not detected in the GT-S strain (27). CTGGGGGAAAAATGTTGGATTGATAACCTCGCCCCGGTTACCATTGAGTCCCATGTGTGTATTTCCCAGGGCGTTTACCTATGCACTGGCAACCACGATTGG |
| 1192983 | SNP | A | A | A | C/A | C/A | C/A | Mummer | slr1855 | slr1855 | T167P | Potential heterogeneous nucleotide (Intensity of the peaks due to C and A were almost equal.) This SNP was not detected in the GT-S strain (27) | |
| 2048341–2049583 | Indel | IS | IS | IS | — | — | — | 99 | BreakDancer | slr1635 | ISY203e | — | Insertion of transposase (14). Specific IS in GT-Kazusa and GT-S strains (27) |
The left column shows the genomic locus of each mutation in the database (NCBI accession number; NC_000911). Quality scores indicate the phred-scaled scores called by MAQ and BWA, respectively. Quality scores given by BreakDancer is a software-original value. The upper table listed the mutations that were suggested as the error of the database and also that the GT-Kazusa strain-specific mutations such as ISY203b, ISY203g, and the locus 2204584 (31). Lower table shows additional differences found only in GT-I, PCC-P and PCC-N strains. Greyed columns emphasize the different sites and their details. Several indel regions miscalled as SNP by MAQ and BWA were shown in parentheses.
Table 3.
List of the genomic loci of SNPs and indels found in the specific strains compared with the nucleotide sequence in the database
| Genomic loci | Type | Data base | GT-Kazusa | GT-S strain | GT-I strain | PCC-P strain | PCC-N strain | Quality score | Source | Gene ID | Annotation | Amino acid change | Comment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 126257 | SNP | C | C | C | C | T | T | 255 255 | MAQ BWA mummer | sll0698 | hik33 | D63N | Different site between GT strains and PCC strains |
| 731367 | Indel | T | T | T | T | — | — | 155 | BWA mummer | sll1574 | sll1574 | sll1574+sll1575 | T insertion in GT strains causes gene split of the original spkA gene. Different site between GT/ATCC strains and PCC strains (12) |
| 781625–781626 | Indel | — | — | — | — | 154 bp | 154 bp | — | — | intergenic region slr2030-slr2031 | slr2030-slr2031 | — | Different site between GT strains and PCC strains (13). TTTAAACGTCATGCACCAATCTCTGATTTACTGGTTTATTCATCTATCAATTCCATAGGCTTTTTGCTTCATCGCTCCAACTAACTTTTCTGGGATGTCCTCCATGCCCCCCGTGCCTAGCTTACCGTCCACCGATGCCGTTATTCCCCCCGGC |
| 831647 | SNP | C | C | C | C | T | T | 255 255 | MAQ BWA mummer | intergenic region ssl3441-sll1815 | infA-adk | — | Different site between GT strains and PCC strains |
| 1204616 | SNP | G | G | G | G | A | A | 255 255 | MAQ BWA mummer | slr1865 | slr1865 | C114Y | Different site between GT strains and PCC strains |
| 1300941–1300985 (1300977) | Indel (SNP) | 45 bp | 45 bp | 45 bp | 45 bp (C) | —(T) | —(T) | (164 255) | (MAQ BWA) | slr1819 | slr1819 | 15 amino acids deletion | Putative PCC strains-specific 45bp deletion without frameshift. This indel region was called as SNP by MAQ and BWA as shown in parentheses. GGGCTATCCTGCGGGATAGCGACATGACCCTGGCCACTCTCCAGG |
| 1423340–1423341 | Indel | — | — | — | — | A | A | 386 | BWA mummer | sll1951 | sll1951 | N1438* Stop codon | Different site between GT strains and PCC strains |
| 1437389 | SNP | A | A | A | A | G | G | 255 255 | MAQ BWA mummer | slr1993 | thl | N6S | Different site between GT strains and PCC strains |
| 1812419 | SNP | C | C | C | C | T | T | 255 255 | MAQ BWA mummer | slr1983 | slr1983 | A225V | Different site between GT strains and PCC strains |
| 2521013 | SNP | T | T | T | T | C | C | 255 255 | MAQ BWA mummer | slr0222 | slr0222 | F898S | Different site between GT strains and PCC strains |
| 2736514–2736515 | Indel | — | — | — | — | T | T | 257 | BWA mummer | sll0182 | sll0182 | Frameshift | Different site between GT strains and PCC strains |
| 3014665 | SNP | T | T | T | T | C | C | 255 255 | MAQ BWA mummer | slr0302 | slr0302 | 92 Silent | Different site between GT strains and PCC strains |
| 3096187 | SNP | T | T | T | T | C | C | 66 | MAQ | ssr1175 | ssr1175 | I47T | Different site between GT strains and PCC strains |
| 3098707 | SNP | T | T | T | T | C | C | 217 224 | MAQ BWA | ssr1176 | ssr1176 | C95R | Different site between GT strains and PCC strains. Potential heterogenous nucleotide (Small T peak was also detected in the PCC strains) |
| Genomic loci | Type | Data base | GT-Kazusa | GT-S strain | GT-I strain | PCC-P strain | PCC-N strain | Quality score | Source | Gene ID | Annotation | Amino acid change | Comment |
| 387006 | SNP | C | C | C | T | C | C | 255 255 | MAQ BWA mummer | slr1085 | slr1085 | P109L | GT-I strain-specific |
| 842060 | SNP | C | C | C | T | C | C | 255 255 | MAQ BWA mummer | sll1799 | rplC | R185Q | GT-I strain-specific |
| 909360 | SNP | C | C | C | T | C | C | 255 255 | MAQ BWA mummer | sll1968 | pmgA | E93K | GT-I strain-specific |
| 1392586 | SNP | T | T | T | C | T | T | 255 255 | MAQ BWA mummer | slr1250 | pstB | L204S | GT-I strain-specific |
| 1470212 | SNP | G | G | G | A | G | G | 255 255 | MAQ BWA mummer | sll1605 | fabZ | R46C | GT-I strain-specific |
| 1764198 | SNP | T | T | T | G | T | T | 234 244 | MAQ BWA mummer | slr1962 | slr1962 | F158C | GT-I strain-specific |
| Genomic loci | Type | Data base | GT-Kazusa | GT-S strain | GT-I strain | PCC-P strain | PCC-N strain | Quality score | Source | Gene ID | Annotation | Amino acid change | Comment |
| 125218 | SNP | G | G | G | G | A | G | 255 255 | MAQ BWA mummer | sll0698 | hik33 | T409M | PCC-P strain-specific |
| 1437136 | SNP | G | G | G | G | A | G | 255 255 | MAQ BWA mummer | slr1992 | slr1992 | 146 Silent | PCC-P strain-specific |
| 2674108 | SNP | C | C | C | C | T | C | 255 255 | MAQ BWA mummer | slr0645 | slr0645 | A3V | PCC-P strain-specific |
| 69849 | SNP | G | G | G | G | G | A | 255 255 | MAQ BWA mummer | slr1119 | slr1119 | R189Q | PCC-N strain-specific |
| 125262–125273 | Indel | 12 bp | 12 bp | 12 bp | 12 bp | 12 bp | — | Mummer | sll0698 | hik33 | Four amino acids deletion | PCC-N strain-specific 12base deletion without frameshift. CTGGGTCAACAT | |
| 1597057 | SNP | T | T | T | T | T | G | 255 255 | MAQ BWA mummer | slr1510 | plsX | V88G | PCC-N strain-specific |
| 1763998 | SNP | G | G | G | G | G | C | 255 255 | MAQ BWA mummer | slr1962 | slr1962 | E91D | PCC-N strain-specific |
| 2370197 | SNP | A | A | A | A | A | G | 255 255 | MAQ BWA mummer | slr0370 | gabD | T306A | PCC-N strain-specific |
| 2580625 | SNP | T | T | T | T | T | A | 255 255 | MAQ BWA mummer | intergenic region ssl0105-sll0063 | ssl0105-sll0063 | — | PCC-N strain-specific |
| 2580626 | SNP | A | A | A | A | A | G | 255 255 | MAQ BWA mummer | intergenic region ssl0105-sll0063 | ssl0105-sll0063 | — | PCC-N strain-specific |
| 2881614–2881615 | Indel | — | — | — | — | — | T | 269 | Bwa | slr0079 | gspE | Frameshift | PCC-N strain-specific |
The left column shows the genomic locus of each mutation in the database (NCBI accession number; NC_000911). Quality scores indicate the phred-scaled scores called by MAQ and BWA, respectively. Greyed columns emphasize the different sites and their details. Several indel regions miscalled as SNP by MAQ and BWA were shown in parentheses.
A part of the indel regions was miss-called as SNPs by the mapping programmes. We aligned the genome sequence of the substrains around the indels (Fig. 3) and found that these indels were located in the middle of two direct repeat- or direct repeat-like sequences. Interestingly, these direct repeats found in the deleted regions were not common sequence. Mapping analyses using de novo assembled contigs (Velvet and MUMmer) help to correct the false-positive SNP/indel-calls made by mapping programmes such as BWA and MAQ (Table 2).
Figure 3.

Alignment of the specific indel regions whose consensus read bases were miss-called. (A) The 154-base deletion in the slr2031 gene13 in the GT strains. (B) The 12-base deletion in the hik33 gene in the PCC-N strain. (C) The 45-base deletion in the slr1819 gene in PCC-P and PCC-N strains. (D) The 102-base deletion in the slr1084 gene in the GT-S and Kazusa strains. Deleted regions were underlined and direct-repeat sequences were emphasized by grey colour. These deleted loci were situated in the middle of the direct-repeat sequences.
Some of the mutations confirmed by the Sanger method were putative heterogeneous SNPs. At these SNP loci, we detected a second peak from the other nucleotide. As the Synechocystis genome is multi-copy, some of the genome copies may harbour heterogeneous SNPs. These loci were not in a multi-repeat region of the genome.
A phylogenetic scheme of the history of the Synechocystis sp. PCC 6803 substrains is presented in Fig. 4. The predicted phylogenetic relationship of various strains indicated that the putative root may exist between the PCC branch and the GT branch. It suggests that all existing substrains do not have the original sequence of this organism isolated in 1968.
Figure 4.

Unrooted tree of phylogenetic relationship of various strains of Synechocystis sp. PCC 6803. Known events are indicated on each branch. The number of mutations in each substrain to the database sequence (Kazusa strain) was indicated. The scale bar indicates the distance of branch corresponding to the number of mutations.
4. Discussion
4.1. Problems associated with the heterogeneity of frozen stocks of bacterial strains
Frozen stocks in culture centres are basically stored as heterogeneous cell groups of the particular bacteria, such as E. coli K-12 MG1655.32 Thus, when aliquots of the frozen cells are thawed, selection bias due to the growth conditions arises; this affects the major genotype of the cells in culture. We posit that the heterogeneity of cells in different laboratories affected the results of genetic research or phenotypic analyses and we suggest that post-genomic research on cyanobacteria in the next generation should be based on resequenced strains as the new reference for each laboratory. In studies on post-genomic science using various mutants, it is better to prepare the frozen stock cells of single colonies one by one and also of the parental cell as soon as possible after the isolation of the mutants. It is also unquestionable that long-term cultivation of the cells on the gels or in the liquid cultures is not appropriate to keep the genotype of the cells.
4.2. Potential factors that affect phenotypic differences in 6803 substrains
We found a small number of substrain-specific mutations in PCC-P and PCC-N strains. It can be expected that a mutation accounts for the difference in the phototactic phenotype of PCC-P and PCC-N strains.19 Furthermore, the 14 mutations found in the GT/Kazusa- and the PCC-P/PCC-N strains suggest that their loci may affect the cell motility of these strains8 or their glucose tolerance.3 Our findings may be useful in functional studies on the gene that is disrupted by SNPs or indels in specific substrains. As Kamei et al.12 or Okamoto et al.14 suggested, the real function of several genes could only be studied in specific substrains which have the original nucleotide sequence without any SNPs, indels, or IS insertions.
4.3. Indels located between two direct repeats result in miss-calls by the mapping programme
Confirmation of our results with the Sanger method revealed that specific deletion patterns were miss-called or undetected at high frequency by mapping analysis of short-read type data. Several deleted regions in the middle of direct-repeat sequences were miss-called as SNPs or undetected (Tables 2 and 3). Alignment of the DNA sequence of the three substrains clearly showed direct repeats on both sides of the deletion locus (Fig. 3). After the deletion event only a single direct repeat sequence remained, leading to the hypothesis that the deletion was due to self-crossover. At present we do not know what mechanisms or factors trigger these deletions, but caution should be exercised when we perform long-term cultivation of cyanobacterial cells. This is also a technical problem of mapping analysis to detect the exact positions of SNPs and indels using massive short-read data of next-generation sequencers.
4.4. Problems raised by heterozygous and homozygous SNPs
In this analysis, we found that the threshold value for SNP detection by BWA and MAQ was more than 60% of read data covering the SNP positions. However, Synechocystis sp. strain PCC6803 has a multi-copy genome,33 suggesting that the genome contains unidentified heterozygous SNPs below the threshold value. It is technically difficult to identify all heterozygous SNPs in the genome; if we lower the threshold, the number of false-positive SNPs increases drastically. Mutations hidden as minor heterozygous SNPs may become major SNPs under specific conditions and affect the cell phenotype, an observation reported by Hihara and Ikeuchi16 and Hihara et al.,17 who studied the pmgA mutant. The active retention of heterogeneity may be a strategy of cyanobacteria for acclimation to environmental changes. Heterogeneous SNPs found in the same locus in several substrains such as the loci 1192983 and 3098707 were the candidates to understand such mechanisms. The detection of minor heterozygous SNPs is a future problem for mapping using massive parallel sequencing.
In this study, we identified a number of differences in the genome sequence of laboratory strains and published sequence data derived from the Kazusa strain. Resequence data on PCC substrains will be useful for considering evolutionary events among Synechocystis intra-species. For the reconstruction of informatics in post-genomic studies on Synechocystis sp. PCC 6803, resequence analysis is effective and represents a powerful genetic strategy to identify potential mutation loci in spontaneous mutants with altered phenotypes.
Supplementary data
Supplementary data is available at www.dnaresearch.oxfordjournals.org.
Funding
This study was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (S0801025).
Supplementary Material
References
- 1.Haselkorn R. Genetic systems in cyanobacteria. Methods Enzymol. 1991;204:418–30. doi: 10.1016/0076-6879(91)04022-g. doi:10.1016/0076-6879(91)04022-G. [DOI] [PubMed] [Google Scholar]
- 2.Porter R.D. DNA transformation. Methods Enzymol. 1988;167:703–12. doi: 10.1016/0076-6879(88)67081-9. doi:10.1016/0076-6879(88)67081-9. [DOI] [PubMed] [Google Scholar]
- 3.Williams J.G.K. Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods in Synechocystis 6803. Methods Enzymol. 1988;167:766–78. doi:10.1016/0076-6879(88)67088-1. [Google Scholar]
- 4.Rippka R., Deruelles J., Waterbury J.B., Herdman M., Stanier R.Y. Genetic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 1979;111:1–61. [Google Scholar]
- 5.Kaneko T., Sato S., Kotani H., et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–36. doi: 10.1093/dnares/3.3.109. doi:10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- 6.Burja A.M., Dhamwichukorn S., Wright P.C. Cyanobacterial postgenomic research and systems biology. Trends Biotechnol. 2003;21:504–11. doi: 10.1016/j.tibtech.2003.08.008. doi:10.1016/j.tibtech.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Stanier R.Y., Kunisawa R., Mandel M., Cohen-Bazire G. Purification and properties of unicellular blue-green algae (order Chroococcales) Bacteriol. Rev. 1971;35:171–205. doi: 10.1128/br.35.2.171-205.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeuchi M., Tabata S. Synechocystis sp. PCC 6803—a useful tool in the study of the genetics of cyanobacteria. Photosynth. Res. 2001;70:73–83. doi: 10.1023/A:1013887908680. doi:10.1023/A:1013887908680. [DOI] [PubMed] [Google Scholar]
- 9.Rippka R., Herdman M. Pasteur Culture Collection of Cyanobacterial Strains in Axenic Culture. vol. I. Paris: Institut Pasteur; 1992. Catalogue of strains. [Google Scholar]
- 10.Smart L.B., McIntosh L. Expression of photosynthesis genes in the cyanobacterium Synechocystis sp. PCC 6803: psaA-psaB and psbA transcripts accumulate in dark-grown cells. Plant Mol. Biol. 1991;17:959–71. doi: 10.1007/BF00037136. doi:10.1007/BF00037136. [DOI] [PubMed] [Google Scholar]
- 11.Kamei A., Ogawa T., Ikeuchi M. Identification of a novel gene (slr2031) involved in high-light resistance in the cyanobacterium Synechocystis sp. PCC 6803. In: Garab G., editor. Photosynthesis: Mechanism and Effects. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1998. pp. 2901–5. [Google Scholar]
- 12.Kamei A., Yuasa T., Orikawa K., Geng X.X., Ikeuchi M. A eukaryotic-type protein kinase, SpkA, is required for normal motility of the unicellular cyanobacterium Synechocystis sp. strain PCC 6803. J. Bacteriol. 2001;183:1505–10. doi: 10.1128/JB.183.5.1505-1510.2001. doi:10.1128/JB.183.5.1505-1510.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katoh A., Sonoda M., Ogawa T. A possible role of 154-base pair nucleotides located upstream of ORF440 on CO2 transport of Synechocystis PCC6803. Photosynthesis: From Light to Biosphere. 1995;3:481–4. Kluwer Academic Publishers: Dordrecht, The Netherlands, [Google Scholar]
- 14.Okamoto S., Ikeuchi M., Ohmori M. Experimental analysis of recently transposed insertion sequences in the cyanobacterium Synechocystis sp. PCC 6803. DNA Res. 1999;6:265–73. doi: 10.1093/dnares/6.5.265. doi:10.1093/dnares/6.5.265. [DOI] [PubMed] [Google Scholar]
- 15.Yoshihara S., Geng X., Okamoto S., et al. Mutational analysis of genes involved in pilus structure, motility and transformation competency in the unicellular motile cyanobacterium Synechocystis sp. PCC 6803. Plant Cell. Physiol. 2001;42:63–73. doi: 10.1093/pcp/pce007. doi:10.1093/pcp/pce007. [DOI] [PubMed] [Google Scholar]
- 16.Hihara Y., Ikeuchi M. Mutation in a novel gene required for photomixotrophic growth leads to enhanced photoautotrophic growth of Synechocystis sp. PCC 6803. Photosynth. Res. 1997;53:129–39. doi:10.1023/A:1005815321295. [Google Scholar]
- 17.Hihara Y., Sonoike K., Ikeuchi M. A novel gene, pmgA, specifically regulates photosystem stoichiometry in the cyanobacterium Synechocystis species PCC 6803 in response to high light. Plant Physiol. 1998;117:1205–16. doi: 10.1104/pp.117.4.1205. doi:10.1104/pp.117.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivatsan A., Han Y., Peng J., et al. High-precision, whole-genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet. 2008;4:e1000139. doi: 10.1371/journal.pgen.1000139. doi:10.1371/journal.pgen.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshihara S., Suzuki F., Fujita H., Geng X.X., Ikeuchi M. Novel putative photoreceptor and regulatory genes Required for the positive phototactic movement of the unicellular motile cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol. 2000;41:1299–304. doi: 10.1093/pcp/pce010. doi:10.1093/pcp/pce010. [DOI] [PubMed] [Google Scholar]
- 20.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. doi:10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. doi:10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H., Handsaker B., Wysoker A., et al. 1000 Genome Project Data Processing Subgroup, The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen K., Wallis J.W., McLellan M.D., et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009;6:677–81. doi: 10.1038/nmeth.1363. doi:10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zerbino D.R., Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2009;18:821–9. doi: 10.1101/gr.074492.107. doi:10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurtz S., Phillippy A., Delcher A.L., et al. Open source MUMmer 3.0 is described in ‘Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. doi:10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sasaki N.V., Sato N. CyanoClust: comparative genome resources of cyanobacteria and plastids. Database (Oxford) 2010 doi: 10.1093/database/bap025. bap025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tajima N., Sato S., Maruyama F., Kaneko T., et al. Genomic structure of the cyanobacterium Synechocystis sp. PCC 6803 strain GT-S. DNA Res. 2011 doi: 10.1093/dnares/dsr026. doi:10.1093/dnares/dsr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsenstein J. PHYLIP—Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–6. [Google Scholar]
- 29.Kanesaki Y., Yamamoto H., Paithoonrangsarid K., et al. Histidine kinases play important roles in the perception and signal transduction of hydrogen peroxide in the cyanobacterium Synechocystis sp. PCC 6803. Plant J. 2007;49:313–24. doi: 10.1111/j.1365-313X.2006.02959.x. doi:10.1111/j.1365-313X.2006.02959.x. [DOI] [PubMed] [Google Scholar]
- 30.Paithoonrangsarid K., Shoumskaya M.A., Kanesaki Y., et al. Five histidine kinases perceive osmotic stress and regulate distinct sets of genes in Synechocystis. J. Biol. Chem. 2004;279:53078–86. doi: 10.1074/jbc.M410162200. doi:10.1074/jbc.M410162200. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki I., Kanesaki Y., Mikami K., Kanehisa M., Murata N. Cold-regulated genes under control of the cold sensor Hik33 in Synechocystis. Mol. Microbiol. 2001;40:235–44. doi: 10.1046/j.1365-2958.2001.02379.x. doi:10.1046/j.1365-2958.2001.02379.x. [DOI] [PubMed] [Google Scholar]
- 32.Nahku R., Peebo K., Valgepea K., Barrick J.E., Adamberg K., Vilu R. Stock culture heterogeneity rather than new mutational variation complicates short-term cell physiology studies of Escherichia coli K-12 MG1655 in continuous culture. Microbiology. 2011;157:2604–10. doi: 10.1099/mic.0.050658-0. doi:10.1099/mic.0.050658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chauvat F., Rouet P., Bottiu H., Boussac A. Mutagenesis by random cloning of an Escherichia coli kanamycin resistance gene into the genome of the cyanobacterium Synechocystis PCC 6803: Selection of mutants defective in photosynthesis. Mol. Gen. Genet. 1989;216:51–9. doi: 10.1007/BF00332230. doi:10.1007/BF00332230. [DOI] [PubMed] [Google Scholar]
- 34.Bhaya D., Bianco N.R., Bryant D., Grossman A. Type IV pilus biogenesis and motility in the cyanobacterium Synechocystis sp. PCC6803. Mol. Microbiol. 2000;37:941–51. doi: 10.1046/j.1365-2958.2000.02068.x. doi:10.1046/j.1365-2958.2000.02068.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.