

Abstract
The malignant transformation of cells requires adaptations across multiple metabolic processes to satisfy the energy required for their increased rate of proliferation. Dysregulation of lipid metabolism has been a hallmark of the malignant phenotype; increased lipid accumulation secondary to changes in the levels of a variety of lipid metabolic enzymes has been documented in a variety of tumors, including prostate. Alterations in prostate lipid metabolism include upregulation of several lipogenic enzymes as well as of enzymes that function to oxidize fatty acids as an energy source. Cholesterol metabolism and phospholipid metabolism are also affected. With respect to lipogenesis, most studies have concentrated on increased expression and activity ofthe de novo fatty acid synthesis enzyme, fatty acid synthase (FASN), with suggestions that FASN might function as an oncogene. A central role for fatty acid oxidation in supplying energy to the prostate cancer cell is supported by the observation that the peroxisomal enzyme, α-methylacyl-CoA racemase (AMACR), which facilitates the transformation of branched chain fatty acids to a form suitable for β-oxidation, is highly overexpressed in prostate cancer compared with normal prostate. Exploitation of the alterations in lipid metabolic pathways in prostate cancer could result in the development of new therapeutic modalities as well as provide candidates for new prognostic and predictive biomarkers. AMACR has already proven to be a valuable biomarker in distinguishing normal from malignant prostate tissue, and is used routinely in clinical practice.
Keywords: Prostate cancer, lipogenesis, fatty acid oxidation
Cancer cell survival requires metabolic alterations
The uncontrolled growth characteristic of malignant transformation requires adaptive alterations in intermediary metabolism to satisfy the energy requirements and biochemical needs of a rapidly dividing cell. Many cancers depend on glycolysis to achieve this goal, a phenomenon first described in the early part of the 20th century by Otto Warburg [1]. He suggested that the increase in glucose utilization via glycolysis was due to an inherent defect in the oxidative phosphorylation mechanism in the malignant cell, and further postulated that this defect in oxidative phosphorylation was a cause of cancer. Although this hypothesis has been proven to be incorrect, in that cancer cells do not have inherent defects in oxidative phosphorylation [2], the observation that cancer cells exhibit increased glycolysis and glucose utilization has proven to be the case for the majority of malignancies, and the Warburg Hypothesis as currently understood refers to the increased glucose utilization characteristic of the malignantstate [3-5]. Furthermore, this increased utilization of the glycolytic pathway can occur even in the presence of adequate oxygen, thus the glycolysis of malignancy has been termed aerobic glycolysis, even though there is no requirement for oxygen. The increase in glucose utilization forms the basis for a diagnostic imaging technique known as positron-emission tomography (PET), in which uptake of a glucose analog is monitored as an indicator of malignancy [6]. Whileenhanced glucose metabolism provides a nutrient source and the energy required for subsequent synthesis of the macromolecular components, such as lipids that are necessary for cellular proliferation, as shown in Figure 1, it has also been noted that there are alterations in the expression of the enzymes that function to facilitate synthesis of these macromolecules as well as in the expression of protein entities, such as growth factors, transcription factors and receptors that are involved in regulating these pathways. There appears to be a global alteration in the metabolome of cancer cells that provides a growth and survival advantage that may be specific to various stages of tumor progression, and may even serve as a the basis for prognostic biomarkers as well as for chemotherapeutic targets [7].
Figure 1.
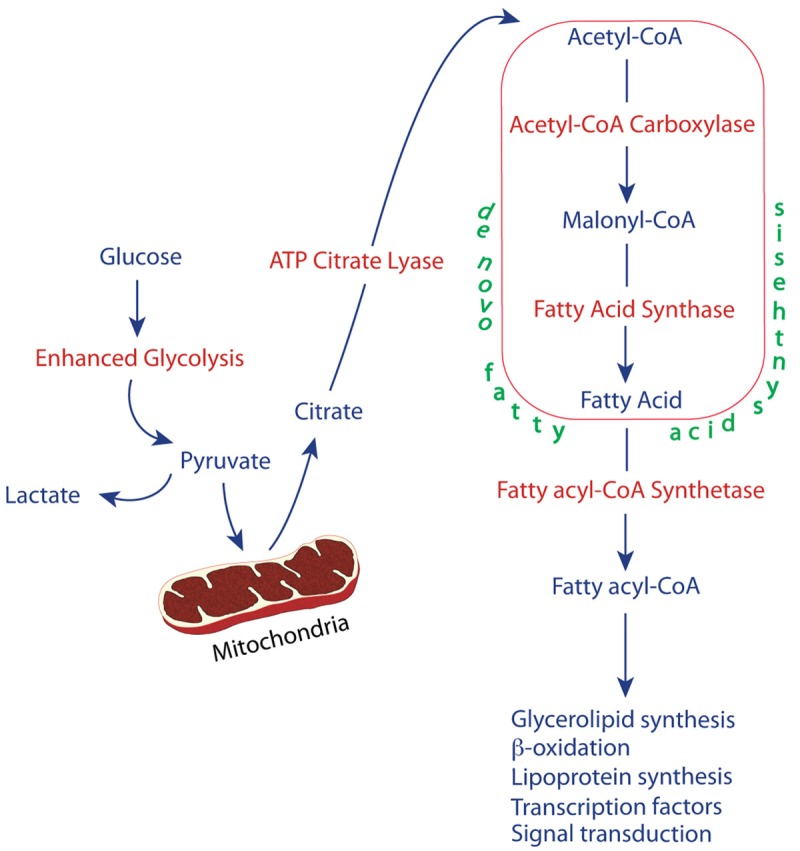
The relationship between enhanced glycolysis and lipogenesis.
Prostate cancer exhibits a unique metabolic adaptation
The normal prostate has a unique intermediary metabolic profile in that it functions as a source of secreted citrate and zinc. The production of large amounts of citrate required for secretion into prostatic fluid is bioenergetically costly, and prostate epithelial cells naturally turn to aerobic glycolysis to compensate for the loss. Thus under normal circumstances prostate cells derive energy via aerobic glycolysis, with less dependence on aerobic oxidation. Malignant prostate cells, on the otherhand, increase levels of oxygen consumption in order to support uncontrolled proliferation. As a result, the large amount of citrate that would normally be secreted, can now function as an intermediate in the citric acid cycle as well as a substrate for de novo fatty acid synthesis [8,9].
Lipid metabolism is important in prostate cancer
The role of lipid metabolism in the development and progression of prostate cancer has been investigated on several levels. While the relationship between obesity and the risk of developing prostate cancer is not yet clear, that between obesity and death from prostate cancer has been more firmly established [10]. And the crucial importance of lipids, including fatty acids, phospholipids and cholesterol in the expression of the malignant phenotype is clear [11,12]. Data suggest that a variety of mechanisms may function to fulfill the need for lipids, including uptake of circulating lipids [13], transfer of fatty acids from stromal adipocytes to prostate cancer cells [14], increased de novo synthesis of fatty acids [15] and phospholipids [16,17], and increased accumulation of cholesterol as cholesteryl ester stored in cytosolic lipid droplets [18]. The role of increased fatty acid oxidation as a source of energy in prostate cancer has been suggested [19], and the profound overexpression of the peroxisomal enzyme, α-methylacyl-CoA racemase (AMACR), that is required for oxidation of branch-chained fatty acids, supports this hypothesis [20]. One can envision a balance between lipid synthesis and oxidation that functions to provide both the lipids and the energy required to support the growth and survival of prostate cancer.
Lipid synthesis is increased in prostate cancer
In most human tissues, the de novo pathway of fatty acid synthesis is of minor importance, the exceptions being liver and mammary gland, and, to a lesser extent, adipose tissue [21]. The cellular requirement for fatty acids is generally met by utilization of dietary fatty acids. Under normal conditions, de novo fatty acid synthesis is suppressed by circulating dietary fatty acids. Cancer cells, however, are insensitive to this product inhibition, and actively synthesize fatty acids despite what would normally be considered an inhibitory level of circulating fatty acids. So, as with many other pathological aberrations, the alteration in de novo fatty acid synthesis in malignancy is likely one of dysregulation. Prostate cancer cells exhibit increased expression of many lipogenic enzymes, often as a result of stimulation by oncogenic signaling pathways such as PI3K/AKT and HER2 [12]. Conversely, increased expression of fatty acid synthetic enzymes has been linked to activation and nuclear localization of Akt/PKB in clinical tumor samples [22]. Androgens have been demonstrated to increase the activity of lipogenic enzymes [23]. A survey of the gene expression database, Oncomine [24], reveals that mRNAs encoding lipid metabolic enzymes, including those involved in de novo fatty acid synthesis (ATP citrate lyase (ACLY), acetyl-CoA carboxylase α (ACACA), fatty acid synthase (FASN), and long chain fatty acyl-CoA synthetases 1, 3 and 5 (ACSL1, ACSL3, ACSL5)) are increased in prostate cancer, as is that of the transcription factor, sterol regulatory binding protein 1 (SREBP1), that regulates expression of fatty acid and cholesterol synthesizing enzymes. In an analysis of published data from multiple separate studies detailed in the Oncomine database, ACLY mRNA expression is significantly increased in 11/22 and decreased in 2/22 studies; ACACA mRNA is increased in 14/21 and decreased in 1/21; FASN mRNA is increased in 12/22 and decreased in 3/22; ACSL1 mRNA is increased in 5/22 and decreased innone; ACSL3 mRNA is increased in 8/20 and decreased in 1/20; and ACSL5 is increased in 6/12 and decreased in none. Expression of SREBP1 mRNA is increased in 8/22 studies and decreased in 2/22. Table 1 summarizes data from several representative studies [25-31] that indicate the fold change and statistical significance of this mRNA overexpression in human tumor samples. The importance of cholesterol metabolism has also recently been reaffirmed. Enhanced cholesteryl ester accumulation in cellular lipid droplets was demonstrated to be associated with prostate cance aggression [18]. In addition, it has been suggested that high circulating levels of cholesterol are positively associated with the development of prostate cancer [32].
Table 1.
Lipogenic enzyme mRNA expression in normal and cancerous human prostate
| Enzyme | Study | Fold Change | p-value | n |
|---|---|---|---|---|
| ACLY | Singh [26] | 2.98 | 1.33e-05 | 50, 52 |
| Welsh [27] | 2.12 | 9.12e-06 | 9, 25 | |
| Wallace [29] | 2.31 | 0.001 | 20, 69 | |
| ACACA | Vanaja [28] | 2.38 | 1.86e-06 | 8, 27 |
| Luo [31] | 1.64 | 0.002 | 15, 15 | |
| Lapointe [30] | 1.72 | 2.31e-13 | 41, 61 | |
| Welsh [27] | 1.46 | 2.80e-06 | 9, 25 | |
| FASN | Singh [26] | 5.64 | 1.96e-06 | 50, 52 |
| Welsh [27] | 3.21 | 4.90e-08 | 9, 25 | |
| Wallace [29] | 3.68 | 1.55e-04 | 20, 69 | |
| Vanaja [28] | 3.20 | 7.65e-05 | 8, 27 | |
| ACSL1 | Singh [26] | 2.21 | 3.45e-05 | 50, 52 |
| Welsh [27] | 1.48 | 0.001 | 9, 25 | |
| Wallace [29] | 1.60 | 0.009 | 20, 69 | |
| ACSL3 | Singh [26] | 1.78 | 1.09e-04 | 50, 52 |
| Lapointe [30] | 1.50 | 1.68e-06 | 41, 62 | |
| Welsh [27] | 1.60 | 5.85e-04 | 9, 25 | |
| ACSL4 | Singh [26] | -1.48 | 0.009 | 50, 52 |
| Welsh [27] | -1.93 | 3.14e-04 | 9, 25 | |
| Grasso [25] | -1.48 | 1.92e-05 | 28, 59 | |
| ACSL5 | Grasso [25] | 2.12 | 8.24e-09 | 28, 59 |
| Lapointe [30] | 1.37 | 1.02e-05 | 41, 60 | |
| SREBP1 | Wallace [29] | 1.53 | 2.41e-04 | 20, 69 |
| Welsh [27] | 1.72 | 4.19e-04 | 9, 25 | |
| Singh [26] | 2.05 | 0.031 | 50, 52 |
Fold change = cancer/normal. n = number of samples (normal, cancer). Data taken from Oncomine [24].
ACLY
As noted in Figure 1, acetyl CoA required for fatty acid synthesis is derived via metabolism of citrate by the enzyme ACLY. Not only is expression of the mRNA that encodes this enzyme increased in prostate cancer, as illustrated in Table 1, but downregulation of its expression in prostate cancer cells either by pharmacologic reagents or siRNA treatment results in inhibition of proliferation [33,34]. Its expression has been reported to be induced by androgen treatment [35].
ACACA
The rate-limiting step in the synthesis of fatty acids is the ATP-dependent conversion of acetyl CoA to malonyl CoA by the enzyme, ACAC. Two isoforms have been identified, ACAC1 (also called ACACA) and ACAC2 (also called ACACB). Physiological control ofACAC activity is mediated by hormones and nutritional status, with SREBP1c playing a major role in regulating ACACA expression [36]. Inhibition of ACACA activity either pharmacologically or by RNA interference resulted in cessation of proliferation and celldeath [37,38]. Like ACLY, ACACA expression is stimulated by androgens [35].
FASN
The most extensively studied of the lipogenic enzymes in the context of carcinogenesis is FASN [39]. Overexpression of this enzyme has been reported for a variety of cancers, including prostate, liver, ovarian, colon, endometrial and breast, and has been associated with malignant transformation and a worse prognosis. In non-malignant tissues, such as liver, FASN activity is regulated by insulin and nutritional status via induction of a family of transcription factors, sterol regulatory element binding proteins (SREBP) that increase the expression of genes involved in lipid metabolism. The FASN of malignancy is non-responsive to nutritional status, and has been shown to be both regulated by [40], and able to regulate [22,41], signal transduction pathwaysthat are associated with the malignant phenotype. It has even been suggested that FASN, itself, functions as an oncogene [42]. Results from a number of studies suggest that FASN might serve as a biomarker of prostate cancer progression, with increased expression associated with a more aggressive phenotype [43,44]. Transgenic expression of FASN in cultured prostate cancer cells increased the rate of proliferation and inhibited apoptosis, while FASN expression in human prostate tumor samples was inverselyassociated with the apoptotic rate [45]. High FASN expression in tumor samples was linked to activation and nuclear localization of Akt [22], and increased FASN was also associated with cytoplasmic stabilization of β-catenin [46]. Downregulation of FASN expression by siRNA, on the other hand, attenuated growth and induced apoptosis [47]. In a separate study [48], FASN inhibition not only suppressed cell proliferation but prevented pseudopodia formation and suppressed cell adhesion, migration, and invasion. FASN inhibition also suppressed genes involved in production of intracellular second messenger arachidonic acid and androgen hormones, both of which promote tumor progression. Androgens have been demonstrated to increase lipid accumulation [49] as well as FASN expression and activity in cultured prostate cancer cells [23]. Androgens have also been shown to increase FASN activity via increasing expression of ubiquitin-specific protease-2a, which, in turn, stabilizes FASN [50]. The 5-alpha reductase inhibitor,dutasteride, reduces FASN mRNA, protein and enzyme activity [51]. As with ACLY and ACACA, pharmacologic inhibition of FASN activity inhibits proliferation and induces cell death in prostate cancer cells, leading to the suggestion that FASN inhibition might be exploited in the treatment of prostate cancer [52]; however, to date, there has not been a viable inhibitor candidate for use in clinical trials due to unacceptable side effects.
Stearoyl-CoA desaturase-1 (SCD1)
The product of FASN activity is predominantly the saturated fatty acid, palmitic acid. Some evidence suggests that the proportion of monounsaturated fatty acids increases in prostate cancer [53], leading to the hypothesis that SCD1 activity may be increased in addition to FASN. Results are equivocal. In 2005, Moore et al [54] reported that SCD mRNA levels were downregulated in prostate cancer relative to normal prostate epithelium. These results were validated by qPCR and immunohistochemical analysis. The median SCD expression levels were 150, 45 and 10 for normal, PIN and carcinoma samples, respectively. However, Fritz et al. reported an increase in both SCD1 mRNA and protein expression in prostate cancer relative to normal prostate, and demonstrated that inhibition of SCD1 activity induces growth arrest of prostate cancer cells in vitro as well as in vivo [53]. A survey of the Oncomine database [24] likewise yields equivocal results: 7/20 analyses show increased mRNA expression in prostatecancer, while 5/20 show decreased expression.
ACSLs
Utilization of fatty acids for either synthesis of neutral and phospholipids or as substrates for β-oxidation requires an activation step catalyzed by a fatty acyl-CoA synthetase (ACS) isoenzyme that converts a free fatty acid to its respective CoA ester. The isoforms are characterized according to the chain length of their preferred substrate. The subset of isoenzymes that act on fatty acids with chain lengths between 16 and 22 carbons are referred to as long chain fatty acyl CoA synthetases (ACSLs).To date, 5 mammalian ACSLs that differ in subcellular location and substrate specificity have been identified [55]. The precise role of each isoform has not been delineated, but current evidence suggests that ACSL1 functions in hepatic glycerolipid synthesis [56,57], while ACSL3 and ACSL5 seem to increase beta oxidation of fatty acids in certain cells [58]. The localization of ACSL4 to peroxisomes suggests that this enzyme may function in fatty acid oxidation [59]. Inhibitor studies indicate that ACSL4 may also be involved in hepatic triacylglycerol synthesis [60]. In prostate, ACSL3 has received the most attention due to its regulation by androgens [61]. While the level of ACSL3 mRNA expression increases in prostate cancer when compared with normalprostate epithelium (Table 1), it appears to be downregulated as a function tumor grade, with high grade and metastatic prostate cancer expressing comparatively lower levels of ACSL3 [61], suggesting a discordance between the growth and differentiation activities of the AR pathway. In prostate cancer cells lines, ACSL3 expression is highest in androgen sensitive cells [62], and treatment of androgen sensitive LNCaP cells with androgens increases expression of ACSL3 mRNA, while decreasing expression of ACSL1 and having no effect on ACSL4, 5, or 6 [63]. Upregulation of ACSL3 by vitamin D3 in LNCaP cells results in growth inhibition [64].
Expression of ACSL4 in prostate cancer cell lines appears to be inversely associated with that of ACSL3 [62], and evidence suggests that ACSL4 expression is associated with androgen-independent growth [65]. Interestingly, ACSL4 is the only isoform that displays decreased expression in cancer versus normal prostate tissue: ACSL4 mRNA expression is decreased in 10/15 analyses and increased in 1/15 (Table 1), yet appears to be a biomarker of androgen resistance and of a more aggressive phenotype of prostate cancer [65]. The same is true for ACSL4 expression in breast cancer [65-68].
Fatty acid oxidation is increased in prostate cancer
Although the emphasis with respect to dysregulation of lipid metabolism in cancer has been on lipogenesis and increased expression of lipogenic enzymes such as FASN, it is clear that fatty acid oxidation also plays a role in supporting the malignant phenotype [69]. A survey of mRNA expression data at Oncomine [24] indicates that a mitochondrial protein that is the rate-limiting step for mitochondrial β-oxidation, CPT1B, is elevated in prostate cancer samples in 12/21 analyses, and decreased in 2/21, while the peroxisomal enzyme that is required for β-oxidation of branched chain fatty acids, AMACR, is elevated in 16/20 analyses, and decreased in none. Representative individual study results are shown in Table 2. The relatively low level of aerobic glycolysis combined with the absence of the glucose transporter, GLUT1 [70], and the overexpression of AMACR [71] as well as of the β-oxidative pathway enzyme, DBP [72], in prostate cancer, support the hypothesis that fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer, as proposed by Liu et al [19]. In addition to its utility as a biomarker, AMACR has also been demonstrated to have functional significiance. Downregulation of AMACR expression via siRNA treatment in prostate cancer cells results in inhibition of proliferation [73], suggesting that AMACR might also serve as a target for therapy of prostate cancer [74].
Table 2.
β-oxidation enzyme mRNA expression in normal and cancerous human prostate
| Enzyme | Study | Fold Change | p-value | n |
|---|---|---|---|---|
| CPT1B | Luo [31] | 11.74 | 4.46e-04 | 15, 15 |
| Wallace [29] | 1.94 | 1.21e-04 | 20, 69 | |
| Welsh [27] | 6.82 | 2.17e-05 | 9, 25 | |
| AMACR | Grasso [25] | 13.05 | 4.57e-24 | 28, 59 |
| Welsh [27] | 10.07 | 2.60e-13 | 9, 25 | |
| Lapointe [30] | 7.22 | 4.11e-23 | 40, 62 | |
| Vanaja [28] | 8.69 | 7.86e-11 | 15, 15 |
Fold change = cancer/normal. n = number of samples (normal, cancer). Data taken from Oncomine [24].
Cholesterol metabolism in prostate cancer
There is ample data supporting a functional relationship between cholesterol metabolism and prostate cancer, ranging from an epidemiologic association between hypercholesterolemia and prostate cancer as well as the efficacy of statins in reducing the risk of prostate cancer, to studies illustrating dysregulation of cholesterol metabolism at the cellular level [32,75]. It has been recently postulated that cholesteryl ester accumulation in lipid droplets within prostate cancer cells is a causative factorunderlying prostate cancer aggressiveness [18]. This aberrant accumulation was demonstrated to be limited to high grade and metastatic disease, and absent from normal tissue, benign prostatic hypertrophy, prostatitis and prostatic intraepithelial neoplasia.Loss of PTEN and subsequent activation of PI3K/Akt/mTOR with upregulation of SREBP and LDL receptors was shown to induce cholesteryl ester accumulation. This effect was independent of androgen signaling. When esterification was blocked at the level of the acetyl-coA cholesterol acyltransferase enzyme, either pharmacologically or by siRNA treatment, there was an observed increase in apoptosis accompanied by a decrease in cellular proliferation, migration and invasion both in vitro and in vivo. These data suggest the centrality of the lipid droplet to regulation of lipid metabolism in cancer. Fatty acids and cholesterol needed to support malignant growth and metastases can be accumulated and stored in these vesicles, and their subsequent mobilization as needed regulated by the phosphorylation state of the perilipin proteins that associate with the lipid droplet membrane.
Summary
The prostate appears uniquely adapted to exploit fatty acid metabolic pathways in support of malignant transformation. The protein changes observed in fatty acid metabolic pathway enzymes could be utilized as biomarkers for prostate cancer diagnosis andprognosis as well as novel targets for treatment. AMACR is now widely used as a biomarker for prostate cancer diagnosis [76,77]. To date, limited efforts to utilize alterations in lipid metabolism to halt uncontrolled growth and metastases have been unproductive, but as evidenced in this review, there are many additional areas to explore as potential targets for treatment. ACSL4 expression, for example, is associated with an aggressive phenotype, while ACSL3 expression is regulated by androgens. Isoform specific inhibitors of either or both these enzymes might abrogate the lipogenic advantage characteristic of prostate cancer cells with minimal general toxicity. A general ACSL inhibitor, triacsin C, has already been demonstrated to be effective against solid tumors in mice [78]. With respect to fatty acid oxidation, many inhibitors are currently available for testing [69].
Acknowledgements
This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (Biomedical Laboratory Research and Development). This study is funded by DoD BCRP Idea Award (BC084403) to MEM, NIH (1U01CA149556-01), DoD PCRP (PC080010 and PC11624) and VA Merit (1I01BX001505-01) grants to PL, NYU Molecular Oncology and Immunology Postdoctoral Training grant (T32 CA009161) to GD.
Disclosure of conflict of interest
None.
References
- 1.Warburg O, Posener K, Negelein E. Uber den stoffwechsel der tumoren. Biochemische Zeitschrift. 1924;152:319–344. [Google Scholar]
- 2.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 5.Hsu PP, Sabatini DM. Cancer Cell Metabolism: Warburg and Beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 6.Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 7.Isidoro A, Casado E, Redondo A, Acebo P, Espinosa E, Alonso AM, Cejas P, Hardisson D, Fresno Vara JA, Belda-Iniesta C, Gonzalez-Baron M, Cuezva JM. Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. Carcinogenesis. 2005;26:2095–2104. doi: 10.1093/carcin/bgi188. [DOI] [PubMed] [Google Scholar]
- 8.Costello LC, Franklin RB. The Intermediary Metabolism of the Prostate: A Key to Understanding the Pathogenesis and Progression of Prostate Malignancy. Oncology. 2000;59:269–282. doi: 10.1159/000012183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mycielska ME, Patel A, Rizaner N, Mazurek MP, Keun H, Patel A, Ganapathy V, Djamgoz MBA. Citrate transport and metabolism in mammalian cells. Bio Essays. 2009;31:10–20. doi: 10.1002/bies.080137. [DOI] [PubMed] [Google Scholar]
- 10.Freedland SJ, Aronson WJ. Obesity and prostate cancer. Urology. 2005;65:433–439. doi: 10.1016/j.urology.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 11.Clarke NW, Brown MD. The Influence of Lipid Metabolism on Prostate Cancer Development and Progression: Is it Time for a Closer Look? Eur Urol. 2007;52:3–4. doi: 10.1016/j.eururo.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 12.Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012;98:1–10. doi: 10.1016/j.prostaglandins.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, Pettus JR, Froehlich HM, Memoli VA, Morganelli PM, Swinnen JV, Timmerman LA, Chaychi L, Fricano CJ, Eisenberg BL, Coleman WB, Kinlaw WB. Lipoprotein Lipase Links Dietary Fat to Solid Tumor Cell Proliferation. Mol Cancer Ther. 2011;10:427–436. doi: 10.1158/1535-7163.MCT-10-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gazi E, Gardner P, Lockyer NP, Hart CA, Brown MD, Clarke NW. Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J Lipid Res. 2007;48:1846–1856. doi: 10.1194/jlr.M700131-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Swinnen JV, Roskams T, Joniau S, Van Poppel H, Oyen R, Baert L, Heyns W, Verhoeven G. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int J Cancer. 2002;98:19–22. doi: 10.1002/ijc.10127. [DOI] [PubMed] [Google Scholar]
- 16.Bertilsson H, Tessem MB, Flatberg A, Viset T, Gribbestad I, Angelsen A, Halgunset J. Changes in Gene Transcription Underlying the Aberrant Citrate and Choline Metabolism in Human Prostate Cancer Samples. Clin Cancer Res. 2012;18:3261–3269. doi: 10.1158/1078-0432.CCR-11-2929. [DOI] [PubMed] [Google Scholar]
- 17.Awwad HM, Geisel J, Obeid R. The role of choline in prostate cancer. Clin Biochem. 2012;45:1548–1553. doi: 10.1016/j.clinbiochem.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, Cheng JX. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab. 2014;19:393–406. doi: 10.1016/j.cmet.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006;9:230–234. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 20.Lloyd MD, Yevglevskis M, Lee GL, Wood PJ, Threadgill MD, Woodman TJ. α-Methylacyl-CoA racemase (AMACR): Metabolic enzyme, drug metabolizer and cancer marker P504S. Prog Lipid Res. 2013;52:220–230. doi: 10.1016/j.plipres.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Weiss L, Hoffman G, Schreiber R, Andres H, Fuchs E, Korber E, Kolb H. Fatty-acid biosynthesis in man, a pathway of minor importance. Purification, optimal assay conditions, and organ distribution of fatty-acid synthase. Biol Chem Hoppe Seyler. 1986;367:905–912. doi: 10.1515/bchm3.1986.367.2.905. [DOI] [PubMed] [Google Scholar]
- 22.Van de Sande T, Roskams T, Lerut E, Joniau S, Van Poppel H, Verhoeven G, Swinnen JV. High-level expression of fatty acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. J Pathol. 2005;206:214–219. doi: 10.1002/path.1760. [DOI] [PubMed] [Google Scholar]
- 23.Swinnen JV, Esquenet M, Goossens K, Heyns W, Verhoeven G. Androgens Stimulate Fatty Acid Synthease in the Human Prostate Cancer Cell Line LNCaP. Cancer Res. 1997;57:1086–1090. [PubMed] [Google Scholar]
- 24.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey T, Chinnaiyan AM. Oncomine: A cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP, Lander ES, Loda M, Kantoff PW, Golub TR, Sellers WR. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–209. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 27.Welsh JB, Sapinoso LM, Su AI, Kern SG, Wang-Rodriguez J, Moskaluk CA, Frierson HF, Hampton GM. Analysis of Gene Expression Identifies Candidate Markers and Pharmacological Targets in Prostate Cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- 28.Vanaja DK, Cheville JC, Iturria SJ, Young CYF. Transcriptional Silencing of Zinc Finger Protein 185 Identified by Expression Profiling Is Associated with Prostate Cancer Progression. Cancer Res. 2003;63:3877–3882. [PubMed] [Google Scholar]
- 29.Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, Stephens RM, Caporaso NE, Loffredo CA, Ambs S. Tumor Immunobiological Differences in Prostate Cancer between African-American and European-American Men. Cancer Res. 2008;68:927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 30.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, Ekman P, DeMarzo AM, Tibshirani R, Botstein D, Brown PO, Brooks JD, Pollack JR. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo JH, Yu YP, Cieply K, Lin F, Deflavia P, Dhir R, Finkelstein S, Michalopoulos G, Becich M. Gene expression analysis of prostate cancers. Mol Carcinog. 2002;33:25–35. doi: 10.1002/mc.10018. [DOI] [PubMed] [Google Scholar]
- 32.Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Curr Opin Pharmacol. 2012;12:751–759. doi: 10.1016/j.coph.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Y, Islam MS, Tian J, Lui VWY, Xiao D. Inactivation of ATP citrate lyase by Cucurbitacin B, a bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett. 2014;349:15–25. doi: 10.1016/j.canlet.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 34.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Swinnen JV, Ulrix W, Heyns W, Verhoeven G. Coordinate regulation of lipogenic gene expression by androgens: Evidence for a cascade mechanism involving sterol regulatory element binding, Äâproteins. Proc Natl Acad Sci U S A. 1997;94:12975–12980. doi: 10.1073/pnas.94.24.12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM. Regulation of acetyl-CoA carboxylase. Biochem Soc Trans. 2006;34:223–227. doi: 10.1042/BST20060223. [DOI] [PubMed] [Google Scholar]
- 37.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical Inhibition of Acetyl-CoA Carboxylase Induces Growth Arrest and Cytotoxicity Selectively in Cancer Cells. Cancer Res. 2007;67:8180–8187. doi: 10.1158/0008-5472.CAN-07-0389. [DOI] [PubMed] [Google Scholar]
- 38.Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA Interference, ÄìMediated Silencing of the Acetyl-CoA-Carboxylase-OE± Gene Induces Growth Inhibition and Apoptosis of Prostate Cancer Cells. Cancer Res. 2005;65:6719–6725. doi: 10.1158/0008-5472.CAN-05-0571. [DOI] [PubMed] [Google Scholar]
- 39.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 40.Van de Sande T, De Schrijver E, Heyns W, Verhoeven G, Swinnen JV. Role of the Phosphatidylinositol 3, Ä≤-Kinase/PTEN/Akt Kinase Pathway in the Overexpression of Fatty Acid Synthase in LNCaP Prostate Cancer Cells. Cancer Res. 2002;62:642–646. [PubMed] [Google Scholar]
- 41.Vazquez-Martin A, Colomer R, Brunet J, Lupu R, Menendez JA. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008;41:59–85. doi: 10.1111/j.1365-2184.2007.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: A metabolic oncogene in prostate cancer? J Cell Biochem. 2004;91:47–53. doi: 10.1002/jcb.10708. [DOI] [PubMed] [Google Scholar]
- 43.Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, Bubley G, Balk S, Loda M. Fatty Acid Synthase Expression Defines Distinct Molecular Signatures in Prostate Cancer1 1 NCI (Director›s Challenge CA84995-04, SPORE in Prostate Cancer CA90381-01A1, and PO1 CA89021-02), Novartis Investigator, and CaPCURE awards. Mole Can Res. 2003;1:707–715. [PubMed] [Google Scholar]
- 44.Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum Pathol. 1996;27:917–921. doi: 10.1016/s0046-8177(96)90218-x. [DOI] [PubMed] [Google Scholar]
- 45.Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, Febbo PG, Subramanian A, Mucci L, Ma J, Signoretti S, Stampfer M, Hahn WC, Finn S, Loda M. Fatty Acid Synthase: A Metabolic Enzyme and Candidate Oncogene in Prostate Cancer. J Natl Cancer Inst. 2009;101:519–532. doi: 10.1093/jnci/djp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fiorentino M, Zadra G, Palescandolo E, Fedele G, Bailey D, Fiore C, Nguyen PL, Migita T, Zamponi R, Di Vizio D, Priolo C, Sharma C, Xie W, Hemler ME, Mucci L, Giovannucci E, Finn S, Loda M. Overexpression of fatty acid synthase is associated with palmitoylation of Wnt1 and cytoplasmic stabilization of [beta] -catenin in prostate cancer. Lab Invest. 2008;88:1340–1348. doi: 10.1038/labinvest.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA Interference-mediated Silencing of the Fatty Acid Synthase Gene Attenuates Growth and Induces Morphological Changes and Apoptosis of LNCaP Prostate Cancer Cells. Cancer Res. 2003;63:3799–3804. [PubMed] [Google Scholar]
- 48.Yoshii Y, Furukawa T, Oyama N, Hasegawa Y, Kiyono Y, Nishii R, Waki A, Tsuji AB, Sogawa C, Wakizaka H, Fukumura T, Yoshii H, Fujibayashi Y, Lewis JS, Saga T. Fatty Acid Synthase Is a Key Target in Multiple Essential Tumor Functions of Prostate Cancer: Uptake of Radiolabeled Acetate as a Predictor of the Targeted Therapy Outcome. PLoS One. 2013;8:e64570. doi: 10.1371/journal.pone.0064570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W, Verhoeven G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology. 1996;137:4468–4474. doi: 10.1210/endo.137.10.8828509. [DOI] [PubMed] [Google Scholar]
- 50.Graner E, Tang D, Rossi S, Baron A, Migita T, Weinstein LJ, Lechpammer M, Huesken D, Zimmermann J, Signoretti S, Loda M. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell. 2004;5:253–261. doi: 10.1016/s1535-6108(04)00055-8. [DOI] [PubMed] [Google Scholar]
- 51.Schmidt LJ, Ballman KV, Tindall DJ. Inhibition of fatty acid synthase activity in prostate cancer cells by dutasteride. Prostate. 2007;67:1111–1120. doi: 10.1002/pros.20602. [DOI] [PubMed] [Google Scholar]
- 52.Kridel SJ, Lowther WT, Pemble IVCW. Fatty acid synthase inhibitors: new directions for oncology. Expert Opin Investig Drugs. 2007;16:1817–1829. doi: 10.1517/13543784.16.11.1817. [DOI] [PubMed] [Google Scholar]
- 53.Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avancès C, Allory Y, de la Taille A, Culine S, Blancou H, Cristol JP, Michel F, Sardet C, Fajas L. Abrogation of De novo Lipogenesis by Stearoyl-CoA Desaturase 1 Inhibition Interferes with Oncogenic Signaling and Blocks Prostate Cancer Progression in Mice. Mol Cancer Ther. 2010;9:1740–1754. doi: 10.1158/1535-7163.MCT-09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore S, Knudsen B, True LD, Hawley S, Etzioni R, Wade C, Gifford D, Coleman I, Nelson PS. Loss of stearoyl-CoA desaturase expression is a frequent event in prostate carcinoma. Int J Cancer. 2005;114:563–571. doi: 10.1002/ijc.20773. [DOI] [PubMed] [Google Scholar]
- 55.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 56.Li LO, Mashek DG, An J, Doughman SD, Newgard CB, Coleman RA. Overexpression of Rat Long Chain Acyl-CoA Synthetase 1 Alters Fatty Acid Metabolism in Rat Primary Hepatocytes. J Biol Chem. 2006;281:37246–37255. doi: 10.1074/jbc.M604427200. [DOI] [PubMed] [Google Scholar]
- 57.Parkes HA, Preston E, Wilks D, Ballesteros M, Carpenter L, Wood L, Kraegen EW, Furler SM, Cooney GJ. Overexpression of acyl-CoA synthetase-1 increases lipid deposition in hepatic (HepG2) cells and rodent liver in vivo. Am J Physiol Endocrinol Metab. 2006;291:E737–744. doi: 10.1152/ajpendo.00112.2006. [DOI] [PubMed] [Google Scholar]
- 58.Zhou Y, Abidi P, Kim A, Chen W, Huang TT, Kraemer FB, Liu J. Transcriptional Activation of Hepatic ACSL3 and ACSL5 by Oncostatin M Reduces Hypertriglyceridemia Through Enhanced {beta}-Oxidation. Arterioscler Thromb Vasc Biol. 2007;27:2198–2205. doi: 10.1161/ATVBAHA.107.148429. [DOI] [PubMed] [Google Scholar]
- 59.Lewin TM, Van Horn CG, Krisans SK, Coleman RA. Rat liver acyl-CoA synthetase 4 is a peripheral- membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch Biochem Biophys. 2002;404:263–270. doi: 10.1016/s0003-9861(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 60.Kim JH, Lewin TM, Coleman RA. Expression and Characterization of Recombinant Rat Acyl-CoA Synthetases 1, 4, and 5. Selective inhibition by triacsin c and thiazolidinedIones. J Biol Chem. 2001;276:24667–24673. doi: 10.1074/jbc.M010793200. [DOI] [PubMed] [Google Scholar]
- 61.Marques RB, Dits NF, Erkens-Schulze S, van Ijcken WFJ, van Weerden WM, Jenster G. Modulation of Androgen Receptor Signaling in Hormonal Therapy-Resistant Prostate Cancer Cell Lines. PLoS One. 2011;6:e23144. doi: 10.1371/journal.pone.0023144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao H, Kim Y, Wang P, Lapointe J, Tibshirani R, Pollack JR, Brooks JD. Genome-wide characterization of gene expression variations and DNA copy number changes in prostate cancer cell lines. Prostate. 2005;63:187–197. doi: 10.1002/pros.20158. [DOI] [PubMed] [Google Scholar]
- 63.Wang Q, Li W, Liu XS, Carroll JS, Jänne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. A Hierarchical Network of Transcription Factors Governs Androgen Receptor-Dependent Prostate Cancer Growth. Mol Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qiao S, Tuohimaa P. The role of long-chain fatty-acid-CoA ligase 3 in vitamin D3 and androgen control of prostate cancer LNCaP cell growth. Biochem Biophys Res Commun. 2004;319:358–368. doi: 10.1016/j.bbrc.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 65.Monaco ME, Creighton CJ, Lee P, Zou X, Topham MK, Stafforini DM. Expression of Long-chain Fatty Acyl-CoA Synthetase 4 in Breast and Prostate Cancers Is Associated with Sex Steroid Hormone Receptor Negativity. Transl Oncol. 2010;3:91–98. doi: 10.1593/tlo.09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu X, Li Y, Wang J, Wen X, Marcus MT, Daniels G, Zhang DY, Ye F, Wang LH, Du X, Adams S, Singh B, Zavadil J, Lee P, Monaco ME. Long Chain Fatty Acyl-CoA Synthetase 4 Is a Biomarker for and Mediator of Hormone Resistance in Human Breast Cancer. PLoS One. 2013;8:e77060. doi: 10.1371/journal.pone.0077060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maloberti PM, Duarte AB, Orlando UD, Pasqualini ME, Solano AR, Lopez-Otin C, Podesta EJ. Functional Interaction between Acyl-CoA Synthetase 4, Lipooxygenases and Cyclooxygenase-2 in the Aggressive Phenotype of Breast Cancer Cells. PLoS One. 2010;5:e15540. doi: 10.1371/journal.pone.0015540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orlando UD, Garona J, Ripoll GV, Maloberti PM, Solano AR, Avagnina A, Gomez DE, Alonso DF, Podesta EJ. The Functional Interaction between Acyl-CoA Synthetase 4, 5-Lipooxygenase and Cyclooxygenase-2 Controls Tumor Growth: A Novel Therapeutic Target. PLoS One. 2012;7:e40794. doi: 10.1371/journal.pone.0040794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chandler JD, Williams ED, Slavin JL, Best JD, Rogers S. Expression and localization of GLUT1 and GLUT12 in prostate carcinoma. Cancer. 2003;97:2035–2042. doi: 10.1002/cncr.11293. [DOI] [PubMed] [Google Scholar]
- 71.Luo J, Zha S, Gage WR, Dunn TA, Hicks JL, Bennett CJ, Ewing CM, Platz EA, Ferdinandusse S, Wanders RJ, Trent JM, Isaacs WB, De Marzo AM. alpha Methylacyl-CoA Racemase: A New Molecular Marker for Prostate Cancer. Cancer Res. 2002;62:2220–2226. [PubMed] [Google Scholar]
- 72.Zha S, Ferdinandusse S, Hicks JL, Denis S, Dunn TA, Wanders RJ, Luo J, De Marzo AM, Isaacs WB. Peroxisomal branched chain fatty acid β-oxidation pathway is upregulated in prostate cancer. Prostate. 2005;63:316–323. doi: 10.1002/pros.20177. [DOI] [PubMed] [Google Scholar]
- 73.Zha S, Ferdinandusse S, Denis S, Wanders RJ, Ewing CM, Luo J, De Marzo AM, Isaacs WB. α-Methylacyl-CoA Racemase as an Androgen-Independent Growth Modifier in Prostate Cancer. Cancer Res. 2003;63:7365–7376. [PubMed] [Google Scholar]
- 74.Carnell AJ, Kirk R, Smith M, McKenna S, Lian LY, Gibson R. Inhibition of Human α-Methylacyl CoA Racemase (AMACR): a Target for Prostate Cancer. ChemMedChem. 2013;8:1643–1647. doi: 10.1002/cmdc.201300179. [DOI] [PubMed] [Google Scholar]
- 75.Freeman MR, Solomon KR. Cholesterol and prostate cancer. J Cell Biochem. 2004;91:54–69. doi: 10.1002/jcb.10724. [DOI] [PubMed] [Google Scholar]
- 76.Hameed O, Humphrey PA. Immunohistochemistry in diagnostic surgical pathology of the prostate. Semin Diagn Pathol. 2005;22:88–104. doi: 10.1053/j.semdp.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 77.Andrews C, Humphrey PA. Utility of ERG versus AMACR expression in diagnosis of minimal adenocarcinoma of the prostate in needle biopsy tissue. Am J Surg Pathol. 2014;38:1007–1012. doi: 10.1097/PAS.0000000000000205. [DOI] [PubMed] [Google Scholar]
- 78.Mashima T, Oh-hara T, Sato S, Mochizuki M, Sugimoto Y, Yamazaki K, Hamada JI, Tada M, Moriuchi T, Ishikawa Y, Kato Y, Tomoda H, Yamori T, Tsuruo T. p53-Defective Tumors With a Functional Apoptosome-Mediated Pathway: A New Therapeutic Target. J Natl Cancer Inst. 2005;97:765–777. doi: 10.1093/jnci/dji133. [DOI] [PubMed] [Google Scholar]