

Abstract
Basal cell carcinomas (BCCs) were essentially a molecular ‘black box’ until some 12 years ago, when identification of a genetic flaw in a rare subset of patients who have a great propensity to develop BCCs pointed to aberrant Hedgehog signalling as the pivotal defect leading to formation of these tumours. This discovery has facilitated a remarkable increase in our understanding of BCC carcinogenesis and has highlighted the carcinogenic role of this developmental pathway when aberrantly activated in adulthood. Importantly, a phase 1 first-in-human trial of a Hedgehog inhibitor has shown real progress in halting and even reversing the growth of these tumours.
The fox knows many pathways, but the hedgehog knows how to cause basal cell carcinomas.
(With apologies to Archilochus, 7th century bc.)
Basal cell carcinomas (BCCs) are keratinocyte tumours that are so named because of their histological resemblance to the cells along the basement membrane — the ‘basal’ layer of the epidermis (FIG. 1). They are the most commonly diagnosed human cancer, at least among persons of European ancestry1. Approximately 750,000 BCCs are treated each year in the United States alone. Despite this high frequency, the death rate from BCCs is extraordinarily low, a reflection perhaps of the excellent care provided by physicians and the fact that these tumours metastasize only extremely rarely. Nonetheless, they can cause significant tissue destruction by local invasion. In total, the cost of care for non-melanoma skin cancers (NMSCs) such as BCCs is the fifth highest for all cancers in the Medicare population in the United States2. Our understanding of their molecular pathogenesis has advanced considerably in the past decade, and indeed these tumours now seem to have become the ‘founding member’ of an expanding group of human cancers in which deregulated Hedgehog (HH) signalling is of vital importance. I review here our current understanding of BCCs, including the environmental and genetic factors that contribute to their development, their molecular pathogenesis, the most obvious unanswered questions about them, and how new understanding might be translated into more effective prevention and treatment.
Figure 1. Histological sections of basal cell carcinoma and squamous cell carcinoma of the skin.

a | Basal cell carcinomas (BCCs) are keratinocyte tumours that are so named because of their histological resemblance to the cells along the basement membrane — the ‘basal’ layer of the epidermis. b | Often BCCs are grouped as non-malignant skin cancer together with squamous cell carcinomas of the skin (shown) and several other less common tumours.
Clinical aspects of BCCs
BCCs classically appear as slow-growing, translucent, elevated lesions on the sun-exposed skin of persons of fair complexion3 (FIG. 2). BCCs occur more commonly in men than in women, and they tend to occur after the age of 50. However, younger people may be developing more BCCs, perhaps correlated with the use of ultraviolet (UV) light sunbeds for cosmetic tanning purposes, especially among younger women4,199. BCCs are often grouped together with skin squamous cell carcinomas (SCCs; see FIG. 1b) and with several other less common tumours as NMSCs. SCCs are considerably more likely to metastasize than are BCCs. Unlike SCCs, which are usually preceded by carcinoma in situ type lesions, BCCs have no detectable precursor lesion. Once individuals have developed a BCC, they have a much higher risk of developing additional BCCs: one estimate based on meta-analysis gives a 44% risk of a second BCC developing within 3 years in patients who have developed their first such tumour5. Usually, however, individual patients develop only one or a few BCCs. The incidence of development of new NMSCs is also high in those with cutaneous SCCs, and the second NMSC tends to be of the same type as the first6.
Figure 2. The cutaneous appearance of basal cell carcinoma.
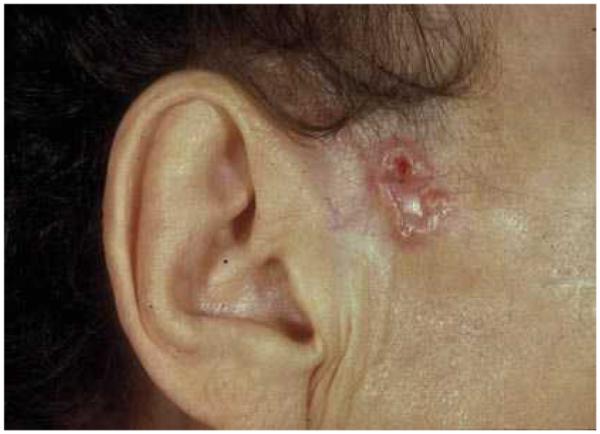
BCCs classically appear as slow-growing, translucent, elevated lesions on the sun-exposed skin of persons with ‘fair’ skin and occur more commonly in men than in women.
BCCs occur far more commonly in persons of European ancestry and in those who have had more sun exposure. Thus, in Kauai, the region of the United States with the highest incidence of skin cancers, the incidence of BCCs is 14-fold higher in persons of European ancestry than in those of Japanese ancestry7 and 34-fold higher than in those of Filipino ancestry8. Among persons of European ancestry, those of Celtic descent are especially prone to developing NMSCs. Patients with albinism, in which constitutional mutations prevent melanin formation, are highly susceptible to the development of BCCs as well as of SCCs and melanoma. By contrast, those of African or South Asian descent with dark skin colour are highly resistant to their development. Interestingly, this marked ethnic difference in susceptibility persists following organ transplantation, when patients become highly susceptible to the development of skin cancers. Thus, the incidence of BCCs may be tenfold higher in organ-transplant recipients of European ancestry than in persons of similar sun exposure who have not had an organ transplant. However, patients of East Asian ancestry have an extremely low incidence of these tumours even after organ transplant9.
As suggested by the inverse correlation of incidence with skin pigmentation, sunlight is an environmental factor that is important in BCC development. However, the exact relationship is complex — apparently far more so than for the development of SCCs. Thus, the incidence of SCCs rises with the total number of hours of sun exposure, especially when that amount approaches a cumulative 100,000 hours. To attain this much sun exposure, one must spend much of the day outdoors. By contrast, the incidence of BCCs peaks at approximately twofold at 10,000–35,000 hours total sun exposure and does not increase with further exposure10,11. Similarly, the relative incidence of SCC:BCC rises with increasing sun exposure — this ratio is considerably higher in the southern parts of the United States than in northern parts. In addition, the use of sunscreens and other sunprotective measures has not yet been found to correlate with reduced BCC risk, unlike the reduction of SCCs associated with sun protection12–14. Some have postulated that BCC development, like that of melanomas, may correlate better with intermittent sunlight exposure, such as that sustained by research oncologists who jog or cycle on weekends15. Additional environmental insults that clearly correlate with BCC development are ionizing radiation and arsenic exposure16–18.
Molecular genetics
BCCs and basal-cell nevus syndrome
The vast majority of BCCs occur sporadically, but there is one rare heritable disorder in which patients have a marked susceptibility to developing BCCs. This is basal-cell nevus syndrome (BCNS, also known as Gorlin syndrome or nevoid basal-cell carcinoma syndrome; see BOX 1).
Box 1. Gorlin syndrome.
Patients with this syndrome, also known as basal cell nevus syndrome (BCNS), had been described previously on numerous occasions but it was the dentist Robert Gorlin who realized most clearly half a century ago that multiple abnormalities occurred in the same patients, thus justifying the ‘syndrome’ designation. This condition is inherited as an autosomal dominant affliction and can produce many, varied phenotypical abnormalities165, most prominent among which are the development of tens, hundreds or even thousands of basal cell carcinomas (BCCs) starting in adolescence or occasionally even in childhood. Patients with BCNS also characteristically develop odontogenic keratocysts, the encroachment of which into the jaws often being the aspect that is most troublesome to the patient. Other tumours to which they are particularly prone are medulloblastomas. Approximately 1–2% of patients with medulloblastomas have BCNS and approximately 3–5% of patients with BCNS develop medulloblastomas, mostly during childhood166. Ovarian fibromas can also occur, sometimes in early childhood or even infancy167. More rarely occurring in BCNS patients, but probably at an incidence greater than in non-BCNS individuals, are meningiomas, rhabdomyosarcomas, cardiac fibromas and mesenteric cysts. The lifespan of patients with BCNS, barring medulloblastomas, seems to be close to normal, but no compilation of their ages or causes of death has been published. Thus, we do not know whether or not these patients have any small increase in incidence of any of the more commonly fatal cancers, including those in which aberrant Hedgehog signalling has been described. Among the more intriguing phenotypical abnormalities in these patients is a strong propensity to develop BCCs after therapeutic ionizing radiation, whether given for treatment of BCCs or of medulloblastomas. BCNS is most often diagnosed on the basis of clinical findings, and major and minor phenotypical criteria for diagnosis have been proposed168.
Using family-based linkage studies of kindreds with BCNS, the locus carrying the causative mutant gene was mapped to human chromosome 9q22 (REF.19) and then to the patched 1 (PTCH1) gene20–22. This finding was especially instructive for several reasons. First, there was previously only minimal insight into the molecular underpinnings of this cancer — p53 mutation in a sizable fraction of sporadic BCCs was essentially the only previously known molecular abnormality23, and p53 mutations are common in non-cancerous skin of BCC patients as well24. This lack of knowledge was due, at least in part, to the difficulty of growing human BCCs in tissue culture or as xenografts and to the lack of a satisfactory animal model. Mice treated with UV or ionizing radiation, or with chemical carcinogens, develop papillomas and carcinomas of the squamous but not basal cell lineage. rats treated with ionizing radiation do develop BCCs but they also develop other skin cancers in greater abundance. Second, the function of PTCH1 was broadly clear — its sequence identified it as the homologue of an already well-studied inhibitor of the HH signalling pathway that was known to be crucial for development in Drosophila melanogaster, and was found subsequently to occupy a similarly crucial role in mammalian development. Hence, it was straightforward to predict and demonstrate that its biallelic inactivation produced constitutive upregulation of HH signalling. Thus, PTCH1 functions as a classic tumour suppressor gene. Third, not only was HH signalling found to be upregulated in all studied sporadic BCCs, but also this upregulation is frequently accompanied by, and at least in part based on, mutations in PTCH1. Currently, it is thought that upregulation of HH signalling is the pivotal abnormality in all BCCs, and indeed there is some evidence that little more than HH upregulation is required for BCC carcinogenesis25,26. Approximately 90% of sporadic BCCs have identifiable mutations in at least one allele of PTCH1 (often loss of the portion of chromosome 9q harbouring PTCH1), and an additional 10% have activating mutations in the downstream smoothened (SMO) protein, which presumably render SMO resistant to inhibition by PTCH1 (REFS 27–30).
Hedgehog signalling
Although a complete description of the intricacies of HH signalling is outside the focus of this review (see REFS 31–33 for reviews), some background information is necessary (FIG. 3). The HH signalling pathway is named after the family of extracellular HH ligands, of which there are three in mammals: sonic hedgehog (SHH), Indian hedgehog (IHH) and desert hedgehog (DHH). PTCH1 is the receptor to which the HH ligands bind, and such binding relieves the inhibition of the pathway induced by unbound PTCH1, specifically through SMO in a non-stoichiometric manner. Once relieved of inhibition, SMO sends signals through a series of interacting proteins, including suppressor of fused (SUFU), culminating in activation of the downstream Gli family of transcription factors, GlI1, GlI2 and GlI3, the founding member of which was identified as a gene amplified in glioblastoma34. These transcription factor proteins exist in various forms, and GlI2 and GlI3 can be activators or suppressors of transcription; GlI1 seems to have only activator functions. The stability of these molecules is controlled by phosphorylation and ubiquitylation–proteolytic destruction, and processing from inactive to active suppressor or activator forms is accomplished, at least in part, by proteolysis35–38.
Figure 3. A basic schematic of the Hedgehog (HH) signalling pathway.

a | The family of extracellular HH ligands, of which there are three in mammals (sonic hedgehog (SHH), Indian hedgehog (IHH) and desert hedgehog (DHH)) bind to the patched 1 (PTCH1) receptor. This relieves the inhibition of smoothened (SMO) by PTCH1, and SMO sends signals through a series of interacting proteins, including suppressor of fused (SUFU), resulting in activation of the downstream Gli family of transcription factors: GLI1, GLI2 and GLI3. b| Loss of PTCH1 in patients with basal cell nevus syndrome predisposes them to basal cell carcinoma (BCC) development. Sporadic BCCs routinely carry mutations in PTCH1 and TP53, consistent with their having been produced by ultraviolet radiation and, in 10% of instances, in SMO. Other mutations have been implicated in BCC development, including genes that regulate skin colour, DNA damage repair genes, members of the phosphoinositide 3-kinase (PI3K)–Akt and the Wnt pathways and FOXM1.
The interactions of the components of the HH signalling machinery can occur at the cilium39–42. SMO seems to be excluded from this structure when inactive, but resides within the cilium when signalling is activated43. Target genes whose expression is upregulated directly by HH signalling in BCCs include PTCH1, providing a negative feedback that dampens of the pathway, GLI1, providing a positive feedback for the pathway, and HHIP, which encodes a HH binding protein44,45. The expression of mrNAs encoding these proteins is routinely increased in BCCs.
Somatic mutations in BCCs
In general, BCCs seem to have relatively stable genomes — the few published studies suggest that they have lower levels of genomic instability than do many extracutaneous cancers46. As noted above, BCCs routinely carry mutations in PTCH1 and TP53 and, in 10% of instances, in SMO47,48. Mutations in several other genes encoding components of HH signalling have been sought but few have been identified, let alone confirmed47. The mutations identified in PTCH1 and TP53 are frequently of a type that is consistent with their having been produced by UV radiation. This is true for BCCs that arise sporadically, and even more so for those large numbers of BCCs that arise in patients with xeroderma pigmentosum (XP), suggesting that repair of UV-induced DNA damage normally does reduce BCC carcinogenesis49–51. Furthermore, this suggests that one reason for the increased incidence of BCCs in older people might be the reported reduction of DNA repair with ageing52.
Predisposing constitutional genetic variants
Because UV irradiation is a significant risk factor for BCC development, genes that control the extent of UV-induced DNA damage and those whose protein products effect repair of that damage are prime candidates for risk-modifying genes.
Variation in constitutive pigment and ability to tan seem to be correlated inversely with BCC development. Hence, it is not surprising that studies have been undertaken of the melanocortin 1 receptor gene (MC1R), the major known genetic variant contributing to the degree of skin pigmentation. MC1R encodes the receptor of α-melanocyte-stimulating hormone (αMSH), a proteolytic product of the larger protein encoded by the pro-opiomelanocortin gene (POMC). MC1R is highly polymorphic among normal individuals and is a prime contributor in determining whether the skin produces brown–black pigment (eumelanin) or red–yellow pigment (pheomelanin). Non-functional MC1r variants result in the production of pheomelanin and, in particular, the phenotype of red hair53. Because people with red hair and fair pigment have an increased risk of skin cancers, those with variant MC1R alleles are at increased risk both of melanomas and of BCCs, and the increased risk is dose-dependent, that is, higher in carriers of two variant alleles than in carriers of a single variant allele54–57. However, in both BCCs and SCCs of the skin, the association of variant alleles and increased risk persists even when corrected for skin pigmentation, thus suggesting a role for MC1R in susceptibility to skin cancers that is mediated by mechanisms other than control of pigmentation. Similarly, MC1R variants seem to contribute to susceptibility to melanoma by mechanisms beyond their effects on skin pigmentation58–60. Indeed, some have suggested that αMSH can directly modulate keratinocyte proliferation and differentiation61. Furthermore, UV irradiation of keratinocytes can enhance POMC and αMSH production, which suggests a possible paracrine role for this hormone62–64. Variation in BCC incidence is also associated with variants in the genes encoding tyrosinase, the rate-limiting enzyme in melanin formation, and agouti signalling protein, which inhibits the interaction of αMSH and MC1r. As with MC1r variants, their effects on pigmentation do not seem to explain all of their effects on BCC susceptibility65.
Patients with XP have constitutional inactivating mutations in both alleles of certain genes that encode DNA repair proteins, in particular those involved in nucleotide excision repair, a process that is of crucial importance for removing UV-induced photoproducts from keratinocyte DNA. In patients with XP, this molecular importance is attested to clinically by the huge increase in relative risk of developing BCCs and other skin cancers and the onset of these cancers on average 50 years earlier than in those without such genetic impairment66.
This finding raises the question of whether more common variants in genes encoding proteins involved in DNA repair might contribute to the relative risk of developing BCCs in patients without XP. Numerous studies have investigated the repair capacities of cells from patients with BCC and control patients, as well as the effects of common coding variants (predominantly single nucleotide polymorphisms) on BCC relative risk. In general, studies in patients with various cancers indicate some reduction of DNA repair67. One group has published several studies that favour a reduced DNA-repair capacity (using the host cell reactivation assay) in lymphocytes from patients with BCC as opposed to controls68, although their findings were not confirmed in a small independent study69.
Similarly, a recent small study found reduced in vivo clearance of UV-induced photoproducts from DNA extracted from the skin of patients with BCC compared with DNA extracted from a control group without skin cancer70. However, the overlap between individuals in the two groups was large, so variations in DNA repair, at least as captured by the assay used, cannot account for a large proportion of any differential susceptibility to BCC in the Finnish population studied.
Associations between DNA repair gene variants and BCC relative risk that reach (or at least closely approach) statistical significance have been reported for several DNA repair genes. Perhaps the most studied of these is the gene polymorphism that underlies the T241M substitution in XRCC3. Some studies71–73 but not all74,75 have found that this substitution is associated with a reduced relative risk for BCCs. Counterintuitively, this substitution is also associated with an increased risk for breast cancer71 and it does not seem to change DNA repair capacity, at least by the assay used. The usual explanation for such findings is that the identified polymorphism is not causative, but is simply located near (is in linkage disequilibrium with) the causative mutation, or it could be that the reported association results are spurious.
Studies of polymorphisms in other DNA repair genes have also suggested associations with the relative risk of developing BCCs, but other studies have failed to find the same association. Clearly, the relationships between identified associations and changes in DNA repair are confusing (BOX 2). Such uncertainties argue for the potential of unbiased, genome-wide association studies to identify polymorphisms that confer a relative susceptibility to BCC formation, and to uncover greater understanding of the mechanisms underlying BCC development.
Box 2. Defects in DNA repair genes that might affect development of basal cell carcinomas.
Studies investigating a possible link between defects in DNA repair genes and basal cell carcinomas (BCCs) have yielded conflicting results. For example, a single nucleotide polymorphism in XPA reported to be associated with increased relative risk of BCCs is also associated with increased, not decreased, DNA repair capacity169; XRCC1 polymorphisms have been reported to be associated170 or not to be associated171 with BCC relative risk; and studies of associations of BCC relative risk with XPD (also known as ERCC2) polymorphisms are in conflict172–175. These disparate results suggest that the effects of any single gene polymorphism may be weak but they do not exclude the possibility of stronger effects of combinations of polymorphisms, and remind us of our lack of knowledge about how the efficiency of DNA repair is controlled in normal individuals. Such controls seem to include not just polymorphisms in the genes encoding these and other DNA repair genes (for example, DNA polymerase-η176), but also potentially polymorphisms in genes encoding other proteins that affect the level of DNA repair, such as interleukin 12, which enhances this process and can apparently affect susceptibility to ultraviolet (UV)-induced skin cancers177. In addition, we have only a partial understanding of the mechanisms by which UV induces skin cancers. These mechanisms probably include not only direct DNA damage but also indirect DNA damage through the production of free radicals and UV-induced immunosuppression178, which may impair putative immune defences against skin cancer development161.
A common variant in TP53 occurs at codon 72, and the two alleles encode either arginine or proline (Pro). Taken together, at least some of the published studies imply that the Pro allele enhances susceptibility to BCC, but perhaps only in people who are relatively more resistant to developing these tumours — those with darker skin pigmentation and a lack of variant MC1R alleles76,77. There are similar published results for an association between this variant and susceptibility to cutaneous melanomas78. Again, as with other studied polymorphisms, conflicting data arguing for no association have also been published79. A further complication of the analysis is an association of the Pro allele with resistance to childhood sunburn. Thus, TP53 alleles may control not only susceptibility to skin cancer but also to sunburn, and the effects of one allele may be opposite on these two phenotypes and in counterintuitive directions. A biologically active polymorphism in MDM2, which influences p53 protein stability, was found not to be associated with BCC relative risk in one study that assessed more than 1,000 subjects80.
One common polymorphism at exon 23 of PTCH1 encodes either Pro or leucine at codon 1315. In patients with more severe BCCs (multiple tumours, early onset), two studies have found an association with the genotype encoding Pro/Pro55,81. However, in one study that was not restricted to patients with greater BCC severity, the individual single nucleotide polymorphism was not associated with BCC relative risk, although one haplotype that included this polymorphism was so associated. Possible associations with the many other genes encoding members of the HH pathway and/or genes encoding controls of this pathway remain to be studied. Finally, Balmain and colleagues reported the surprising finding that a Ptch1 polymorphism in the mouse is an important controlling factor in the susceptibility to mutant HRAS-induced skin tumours of the squamous lineage. They suggest that such polymorphisms may control the relative susceptibility to formation of SCCs versus BCCs82.
Molecular analyses are therefore consistent with the idea of genetic control of susceptibility versus resistance to BCC carcinogenesis. In particular, such analyses are consistent with the idea that resistance is associated with better protection against mutagenesis and with better repair of whatever DNA damage does occur. However, we have yet to identify convincingly the genetic underpinnings beneath the wide range of clinical outcomes in some people of northern European ancestry with pale complexions who sunbathe: some get many BCCs, some get many SCCs, some get fewer tumours, some get only precancerous carcinomas in situ, some get only wrinkles and some sustain no clinically apparent skin damage at all.
Pathway interactions and expression changes
Several groups have assessed genome-wide expression in keratinocytes in which HH signalling has been activated experimentally83 and directly in BCCs. A question with the latter is the choice of control cells against which the BCC expression results are compared. Published studies have compared expression patterns of whole tumour with those of whole normal skin84–86 or of cells at the periphery of BCC nests with those of cells of the basal layer of the interfollicular epidermis87. Overall, perhaps as expected, many differences in expression have been found, but so far the field has not progressed sufficiently to enable firm conclusions to be drawn about which findings are reproducible across different platforms and investigators, and which changes are crucial to the aberrant behaviour of the BCC cancer cells.
Currently, the best-studied downstream mediators remain those on which investigators have chosen to focus as candidate targets. Among these are upregulated plateletderived growth factor receptor-α (PDGFRα)88, the upregulated apoptosis inhibitors BCl2 (REFS 89,90) and CASP8 and FADD-like apoptosis regulator (CFlAR)91 and the downregulated apoptosis inducers CD95 (FAS)92,93 and BMI1 (REFS 94,95). The ‘wiring’ downstream of HH signalling activation seems to differ in various tissues. Thus, inhibition of mitogen-activated protein kinase signalling inhibits the growth of HH pathway-stimulated BCC cells but not that of cerebellar granule precursor cells88,96. But, in truth, we have only a limited understanding of which downstream expression changes are actually crucial for HH-induced BCC carcinogenesis. More data have been published during the past few years indicating that other signalling pathways may have profound effects on HH signalling in cancers (see below and BOX 3), although most of these studies did not address BCCs specifically.
Box 3. Signalling pathways implicated in HH-induced tumours.
Phorbol esters, which activate protein kinase C (PKC), are the most extensively studied promoters of squamous cell carcinogenesis, so it is not surprising that members of the PKC family have been investigated in relation to basal cell carcinomas (BCCs). Initial reports indicated that although PKCα is expressed in the epidermal and hair follicle basal layers, it is not expressed in BCC tumour nests179,180. In model cell systems in vitro, PKCα inhibits the activity of the Gli family of transcription factors. By contrast, PKCδ increases Gli activity, and sonic hedgehog (SHH) activation of Gli in at least some contexts seems to require PKCδ activity181,182. The effects of PKCδ seem to be downstream of suppressor of fused (SUFU) but upstream of GLI1 (REF. 182). PKC also may act through activation of the MAPK kinase (MEK) to control Gli transcriptional activity itself. Like PKCα, PKCδ seems not to be expressed in BCC tumour nests but both are expressed in BCC stroma180. One of the cancer-stimulatory genes downstream of PKC is ornithine decarboxylase (ODC), and in the Ptch1+/− mouse model, ODC inhibitors reduce UV-induced BCC carcinogenesis and reduce expression of hedgehog target genes183. However, ODC activity can be controlled by other pathways as well184.
Epidermal growth factor receptor185–190 and transforming growth factor receptor β191–196 pathways can influence HH signalling markedly in model systems, but there is little evidence for their possible roles in BCC carcinogenesis. Surprisingly, because Notch activation can drive some human leukaemias, Notch loss in mice can activate GLI2 expression and produce skin tumours, including BCCs197,198.
PI3K–Akt
Interactions of the HH signalling pathway and the phosphoinositide 3-kinase (PI3K)–Akt pathway are suggested by models in which HH-induced tumorigenesis is enhanced by concomitant PI3K–Akt signalling activation97 or responses to HH ligands are enhanced by concomitant insulin-like growth factor (IGF) ligand. Indeed, Ptch1+/−Igf2−/− mice fail to develop rhabdomyosarcomas98, but no data about the dependence of BCC formation on Akt signalling have been published. These two classical pathways may interact at several levels. First, activation of HH signalling in some systems can affect PI3K–Akt signalling99–102. Second, PI3K−Akt signalling can affect HH signalling: activated PI3K–Akt can stabilize GlI2 through inhibition of protein kinase A (PKA)-mediated phopshorylation that normally results in ubiquitin-targeting and degradation103.
FOXM1
FOXM1, which encodes a member of the forkhead box of transcription factors, is expressed in BCCs at higher levels than in normal keratinocytes104. This gene is more generally expressed in all proliferating cells, its expression is higher in transformed cell lines and its overexpression contributes to carcinogenesis and to more malignant behaviour in various cancer models105–109. Its transcriptional activity can be activated by DNA damage through CHK2 phosphorylation and consequent protein stabilization and, in turn, its expression stimulates the expression of the DNA repair enzymes XrCC1 and BrCA2 (REF. 110). FOXM1 is a HH target gene, and its loss rescues entry into mitosis in cells specifically driven by HH signalling111. Expression of FOXM1 is crucial for normal mitosis, and its loss can cause chromosomal instability, mitotic catastrophe and consequent cell death112. FOXM1 transcriptional activity can be inhibited by ARF113, and pharmacological inhibition of FOXM1 with a cell-penetrating ARF peptide114 or with a small molecule115 has an anticancer effect in model systems. We do not yet know the degree of dependence of BCCs on FOXM1 expression.
In addition, another FOX family member, FOXE1, is expressed in human epidermis and at a higher level in human BCCs116. Its loss is associated with abnormal development of the hair follicle in a pattern consistent with it being a HH target gene and with it mediating downstream effects of HH signalling117. Again, its downstream target genes and the role it has in BCC carcinogenesis are unknown.
Wnt signalling
Although previous studies have drawn conflicting conclusions regarding the role of Wnt signalling in BCC carcinogenesis, a recent study presents convincing evidence for the requirement for activated Wnt signalling to be downstream of HH signalling in these tumours, in both mice and humans118. These findings suggest yet another target for therapeutic intervention.
In summary, we still have only a dim idea of the factors that control BCC keratinocyte HH signalling, other than driver mutations, and of the ‘wiring’ downstream of this pathway. Often impressive results in model systems suggest that such knowledge could point to potential targets for therapies, which could perhaps be useful in combination with the specific HH inhibitors now under development, especially because modulators of these other pathways are also under development. Hence, a global assessment of these in BCCs remains a high priority.
Mouse models of BCC carcinogenesis
As discussed earlier, classical mouse skin carcinogenesis models readily produce tumours of the squamous lineage but none of the BCC lineage. The identification of the pivotal role of HH signalling in BCC carcinogenesis stimulated the engineering of several models in which HH signalling could be manipulated and BCCs could be produced, some of which carry inactivating mutations in genes encoding inhibitors of HH signalling, and some of which carry activated mutants or overexpressed wild-type positive regulators of this pathway (TABLE 1). These models have allowed studies of interventions — chemoprevention and chemotherapy — as well as more basic investigations into BCC tumorigenesis.
Table 1.
Occurrence of basal cell carcinoma (BCC) in mouse models
| Gene | BCC as a result of overexpression (transgenic) |
BCC as a result of knockout |
|---|---|---|
|
| ||
| Shh | Yes | No |
|
| ||
| Ptch1 | No | Yes |
|
| ||
| Smo | Yes | No |
|
| ||
| Sufu | No | Yes |
|
| ||
| Gli1 | Yes | No |
|
| ||
| Gli2 | Yes | No |
The first lesson derived from the models is that deregulated HH signalling is indeed crucial to BCC carcinogenesis. Thus, either constitutive or conditional overexpression of GlI1 (REF. 119) or of GlI2 (REF. 120) in keratinocytes can produce BCC-like proliferations in the skin. Similarly, expression of SMO carrying the activating mutations identified in human BCCs also can produce murine BCCs29. Furthermore, Ptch1+/− mice develop BCCs, and those BCCs often have deletion of the wild-type copy of Ptch1 as well as upregulation of HH signalling121. Similarly, mice carrying one inactivated, mutant allele of the HH suppressor Sufu are also susceptible to BCC development122. Thus, mice carrying mutations in genes that encode at least four different components of the HH signalling machinery develop BCCs, or at least skin tumours resembling BCCs.
The second lesson is that it seems that the degree of activation of HH signalling is correlated with the histological appearance — the stronger the activation, the more the tumours resemble human BCCs123. With weaker activation, the tumours more closely resemble human tumours that are more hair-follicle-like.
A third striking finding is that p53 loss markedly enhances HH-driven tumorigenesis. This was shown first by the development of medulloblastomas in almost 100% of Ptch1+/−;Trp53−/− mice as opposed to an incidence of less than 10% in Ptch1+/− mice that have wild-type p53 (REF. 124). Similarly, we have found that Ptch1+/− mice in which p53 is deleted conditionally in K14-expressing keratinocytes have a marked enhancement of BCC carcinogenesis. Therefore, the high incidence of p53 mutations in human BCCs is probably not simply caused by the fact that BCCs usually arise in sunexposed skin, but rather reflects the ability of p53 loss to contribute to the development of BCCs and perhaps to that of other HH-driven tumours as well.
Finally, results of pharmacological interventions in the Ptch1+/− mouse seem so far to correlate well with results of the same interventions in humans. For example, topical application of the retinoid tazarotene, a retinoic acid receptor-β/γ ligand that is widely used for treatment of acne, (see later) inhibits BCC development in the Ptch1+/− mouse and has clear anti-BCC efficacy in humans. Similarly, we have found that systemic non-steroidal anti-inflammatory drugs, such as celecoxib, weakly inhibit BCC carcinogenesis in both human and mouse PTCH1+/− individuals (J. Tang et al., unpublished observations).
Interventions
Prevention
Although the use of sunscreens has not so far been associated with a reduction in BCCs, there is hope that their use earlier in childhood might reduce later BCC carcinogenesis. Clinical trials addressing this hypothesis will not be completed in the near future, and so counselling of sun avoidance must currently rely on ‘best guesses’. One alternative to sun-protection measures is that of artificial tanning by systemic administration of αMSH125, and indeed tanned skin is less susceptible to UV-induced DNA damage126. As compared with UV-induced tanning, αMSH-induced tanning would seem to have the advantages of being non-mutagenic, non-promoting and non-immunosuppressive, but its clinical practicality has yet to be demonstrated, and current development efforts involve parenteral drugs, a route of administration that is not likely to gain universal acceptance for UV protection.
Oligonucleotides that mimic the free ends of telomeres enhance DNA repair, stimulate tanning127 and have anticancer efficacy in various cancer models, including UV-treated Ptch1+/− mice128. Another strategy to enhance DNA repair capacities is to apply extra copies of DNA repair enzymes. Surprisingly, such proteins applied in liposomes can penetrate skin and accumulate in the nucleus of UV-damaged cells in an apparently catalytically active form such that significant reduction in UV-induced skin changes, including experimental carcinogenesis, can be seen129. Such topical applications over one year significantly reduced the development of new skin cancers in patients with XP130.
Systemic retinoids have been reported to be effective against new BCC development in patients with BCNS131,132 or XP133, but have shown no protective effect in trials in patients who are at high risk of developing sporadic BCCs134,135. Surprisingly, prolonged topical application of tazarotene can cure 25—50% of sporadic human BCCs136–138. Topical tazarotene effectively prevents BCC carcinogenesis in the Ptch1+/− mouse139,140, and we (D. Bickers and E.H.E.) are now conducting a multicentre clinical trial of its efficacy in patients with BCNS. Finally, one clinical trial of dietary intervention in patients who were at high risk of developing new sporadic skin cancers found that a low-fat diet (20% of calories as fat as opposed to the usual 35—40%) was associated with a reduction in the number of new BCCs141.
Treatment —narrowing the hedgehog’s powers
Surgical excision of BCCs is currently by far the most commonly used treatment, and control approaches 100%. However, this high cure rate is accompanied by the inevitable discomfort and scarring of surgery, and the high frequency of BCCs makes them an expensive effort for those who foot the bill. As PDGFrα has been reported to mediate some of the downstream effects of HH signalling in BCCs, use of approved agents that inhibit this receptor kinase would be sensible88. These include sorafenib and imatinib (Gleevec) but no study of their efficacy in BCC has been published. One systemic chemotherapy reported to have some efficacy for locally uncontrollable BCCs is the combination of paclitaxel and carboplatin142. With the evidence for HH activation in many types of visceral cancers, the rationale for development of HH inhibitors (HHIs) has become compelling. Consequently, at least half a dozen pharmaceutical companies have embarked on HHI development, and BCCs that are not controllable by local therapies are potential targets for trials of HHIs.
The first well-studied HHI is the plant alkaloid cyclopamine143. Indeed, one intrepid group applied cyclopamine topically and reported regression of four sporadic BCCs144. Cyclopamine is a competitive inhibitor of SMO signalling, binding directly to the protein145–147, and inhibits the growth of malignant cells driven by HH activation148. Infinity Pharmaceuticals in Cambridge, Massachusetts, USA is developing cyclopamine derivatives with better pharmacological and inhibitory properties as potential HHIs, and the company expects to launch a phase 1 trial of one of these in 2008.
The first HHI tested in phase 1 trials was a Curis–Genentech compound (Curis 61414), which produced neither clinical changes in the tumours nor reductions of mrNA encoding the HH target gene GLI1 when applied topically to sporadic human BCCs (see Curis website 2006 press release in Further information). However, GDC-0449, a second Curis–Genentech HHI molecule, seemed to have minimal toxicity and to cause clinically significant benefit in eight out of nine patients with metastatic or locally advanced BCCs when administered orally in a phase 1 trial149. Genentech expects shortly to initiate phase 2 trials of this molecule in patients with advanced BCCs and in patients with advanced colorectal and ovarian cancers.
The practical barriers to the development and use of HHIs in localized BCCs include the high cure rate with surgery, despite the unattractive aspects of such procedures described earlier. If systemic delivery of HHI were to be used, the agent would have to be essentially 100% free of adverse extracutaneous effects. One potential on-target adverse effect might be on normal tissue stem cells such as those of the brain, the maintenance of which in mice is supported by HH150,151. loss of these brain stem cells in mice has been reported to give cognitive defects152. Indeed, the minimally annoying dysgeusia (distortion or loss of the sensation of taste) and alopecia seen in the systemic HHI trial could be caused by on-target effects of inhibition of HH function normally required for maintenance of the olfactory bulb, tongue papillae and hair follicle150,153–155. Such therapy-induced loss occurring in humans with pancreatic cancer might be tolerable; not so for patients with the usual BCCs. HHIs also can cause premature closure of epiphyses156, a reminder that development is not complete at birth and a potential contraindication to HHI treatment of childhood medulloblastomas.
Unanswered questions
Why are BCCs so much more common than other human cancers?
The answer to this question traditionally has been that they arise in an organ that is subject to enormous mutagenic insults exogenously and endogenously as a ‘cost’ of the continued lifelong high turnover of the epidermis. Viewed in molecular terms, it might also be the case that, unlike in the development of many visceral cancers, disruption of merely one anticancer mechanism — restraint of HH signalling — seems to be enough to allow their growth. However, viewed in another context (prostate cancer in the elderly, for example), perhaps their frequency is not so high after all. Moreover, the apparently high incidence might be, in part, a product of the fact that these lesions are obvious to the naked eye. Indeed, as we have been able to examine internal tissues more closely, the incidence of ‘incidentalomas’ rises dramatically, with the accompanying quandary of what is the proper medical intervention. If the diagnosis of BCCs were to depend on organ dysfunction rather than appearance, their incidence might plummet to less than that of many classical visceral cancers. In this context, the skin may serve as a useful model for how medicine will learn to deal with the increasing numbers of ‘incidentalomas’ that our improving diagnostic acumen uncovers.
Why does the incidence of BCCs not rise proportionally to the amount of UV exposure as in SCCs?
One possibility for this comes from the observation that sun exposure activates DNA repair mechanisms157: perhaps mutations of PTCH1 are more susceptible to repair than mutations in the genes that underlie SCCs. However, no evidence for such differential repair is available. One alternative is suggested by the observation that vitamin D inhibits HH signalling through binding to SMO protein158. Thus, more frequent UV exposure might maintain a level of keratinocyte vitamin D that is sufficient to inhibit the growth of BCCs but not of SCCs. A potential flaw in this is that one study suggested that chronic sun exposure fails to increase cutaneous vitamin D levels even while increasing internal levels159. Consistent with this idea, however, is the finding that skin production of 7-dehydrocholesterol, the precursor molecule that UV radiation converts to vitamin D, wanes in the elderly, and this parallels the increasing incidence of BCCs with ageing160. More generally, the increasingly studied and discussed anticancer effects of vitamin D stores might also have some anti-BCC carcinogenesis effect. Another possibility might be that the photoimmunosuppression that occurs with sun exposure affects SCCs more than BCCs; indeed, in immunosuppressed organ transplant patients, the incidence of SCCs increases considerably more than the incidence of BCCs. However, at least in Ptch1+/− mice, anti-rejection drugs can allow more robust BCC carcinogenesis161, and so the immune system ordinarily may provide at least some protection against BCC as well as SCC carcinogenesis. Better understanding might come were we able to identify clinically inapparent precursor lesions.
Why do BCCs so rarely metastasize?
Not only do BCCs essentially never spread to distant regions, but also the limitation of their growth to that by local extension allows careful, microscopically controlled surgical excision to give a cure rate approaching 100%. One possibility might be that physicians remove BCCs before they grow to a size that allows the accumulation of the additional mutations needed to metastasize. Yet patients with BCNS may have hundreds of BCCs; commonly, BCCs in these patients are removed only when they impinge on sensitive structures such as the eye, and most are treated with a ‘wait and see’ approach. Another possibility would be that activated HH signalling is incompatible with metastasis. But prostate cancer metastases have higher HH signalling activity than do the primaries from which they arose162, suggesting that this might not be the answer. A third possibility might be that specialized abnormal stroma is required, and indeed there is some evidence that BCCs are especially dependent on stroma, at least for their experimental transplantation to other sites in humans163. In addition, loss of specialized stroma might be one reason for the marked downregulation of HH signalling when experimental BCCs or medulloblastomas are transferred from the host to tissue culture. But in most patients with BCCs, there would seem to be no lack of UV-damaged stroma available as a ‘soil’ for BCCs to find if this were the main cause of their lack of metastasis. Finally, BCCs tend to have relative genomic stability, and perhaps it is this that provides the barrier to further DNA abnormalities that might confer metastatic potential. But why do BCCs not acquire genomic instability? Might such instability be incompatible with their successful local growth?
What can we learn about BCCs that may further inform our knowledge of visceral cancers?
If hedgehog activation at first induces keratinocyte senescence, as has been suggested in experimental models of HH signalling activation164, why does senescence not occur with physiological HH signalling during development, hair follicle cycling and experimental models with conditional Gli overexpression?25 A greater understanding of the HH pathway and the induction of senescence is needed to address this question. It is possible that the degree of deregulation of HH signalling found in human cancer is enough to overcome induction of senescence. Indeed, it will be interesting to determine how much HH signalling is required to drive cancer development in different tissues. Analysis of tissues from the recent phase 1 clinical trials of HHIs in patients with BCC will also provide important data on whether a complete absence of HH signalling is required for a clinical response or whether a reduction is all that is needed. later stage trials of HHIs in BCC will also be able to address whether relapse or resistance occur in patients with advanced BCC. Finding mechanisms of resistance, should they occur, might help the development of HHIs for other cancers such as medulloblastoma, in which HH signalling is deregulated. As BCCs seem to have relatively little genomic instability, it is reasonable to hope that mutations may not be as frequent a mechanism of resistance as is the case with more genetically unstable cancers. Finally, it will be important to establish the signalling pathways downstream of the Gli transcription factors that drive carcinogenesis. These might provide opportunities for development of drugs with perhaps a better therapeutic index (for example, sparing stem cells that, at least theoretically, might be the target of collateral damage with HH inhibition) than that of direct inhibition of HH signalling.
Conclusions
In little more than one decade, thanks to the work of many investigators, our understanding of the molecular pathogenesis of BCCs has gone from close to zero to a fairly significant body of information. Certainly, we still have only a rudimentary knowledge of the genetic underpinnings that determine which people develop BCCs and which do not, and of the wiring that drives keratinocytes to BCC carcinogenesis. But it does seem that we already have enough information to have a reasonable chance of translating our molecular understanding to real clinical benefit. The successful molecularly targeted treatment of BCCs with HHI might be only the first of the clinical benefits that result from the vanquishing of the evil hedgehog.
Medicare.
The US federal government medical insurance programme that covers all citizens over the age of 65.
At a glance.
Basal cell carcinomas (BCCs) are keratinocyte tumours that resemble the basal layer of the epidermis, and are the most commonly diagnosed human cancer among persons of European ancestry.
Despite this high frequency, the death rate is extraordinarily low, a reflection perhaps of the excellent care provided by physicians and of their vanishingly rare propensity to metastasize.
The vast majority of BCCs occur sporadically, but patients with the rare heritable disorder basal cell nevus syndrome (BCNS) have a marked susceptibility to developing BCCs.
Family based linkage studies of kindreds with BCNS identified the patched 1 (PTCH1) gene, an inhibitor of the hedgehog signalling pathway, as being mutated in these patients. p53 is also mutated in some patients with sporadic BCCs.
Downstream signalling pathways that are deregulated in patients with BCCs are currently being investigated.
Surgery is curative for most patients with BCCs. However, for those few that develop locally advanced or metastatic BCC, for which there is currently no effective treatment, Phase I clinical trials with inhibitors of the hedgehog signalling pathway have produced promising results.
Linkage disequilibrium.
When alleles at two or more genetic loci occur more frequently in the population than expected given the known allele frequencies and recombination fraction between the two loci. This indicates that the loci are tightly linked; that is, sufficiently close together on the same chromosome to be co-inherited more than 50% of the time.
Parenteral.
Administration of a drug by injection, such as subcutaneous, intramuscular or intravenous, rather than administration through the alimentary canal.
Cyclopamine.
The teratogenic component of corn lilies that is responsible for the cyclopean (one-eyed) phenotype of lambs born of dams eating this plant.
Acknowledgments
Original research of Ervin H. Epstein is supported by grants from the National Institutes of Health (CA109584, CA115992, AR050440) as well as generous support from the Michael J. Rainen Family Foundation.
Footnotes
Competing interests statement
The author declares competing financial interests: see web version for details.
References
- 1.Miller DL. Nonmelanoma skin cancer in the United States: incidence. J. Am. Acad. Dermatol. 1994;30:774–778. doi: 10.1016/s0190-9622(08)81509-5. [DOI] [PubMed] [Google Scholar]
- 2.Housman TS, et al. Skin cancer is among the most costly of all cancers to treat for the Medicare population. J. Am. Acad. Dermatol. 2003:425–429. doi: 10.1067/mjd.2003.186. [DOI] [PubMed] [Google Scholar]
- 3.Rubin AI, Chen EH, Ratner D, carcinoma Basal-cell. N. Eng. J. Med. 2005;353:2262–2269. doi: 10.1056/NEJMra044151. This paper gives an authoritative, more complete review of the more clinical aspects of this tumour. [DOI] [PubMed] [Google Scholar]
- 4.Karagas MR, et al. Use of tanning devices and risk of basal cell and squamous cell skin cancers. J. Natl Cancer Inst. 2002:224–226. doi: 10.1093/jnci/94.3.224. [DOI] [PubMed] [Google Scholar]
- 5.Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Arch. Dermatol. 2000;136:1524–1530. doi: 10.1001/archderm.136.12.1524. [DOI] [PubMed] [Google Scholar]
- 6.Karagas MR, et al. Risk of subsequent basal cell carcinoma and squamous cell carcinoma of the skin among patients with prior skin cancer. JAMA. 1992;267:3305–3310. [PubMed] [Google Scholar]
- 7.Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Nonmelanoma skin cancer in Japanese ethnic Hawaiians in Kauai, Hawaii: an incidence report. J. Am. Acad. Dermatol. 1995;33:422–426. doi: 10.1016/0190-9622(95)91387-4. [DOI] [PubMed] [Google Scholar]
- 8.Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Non-melanoma skin cancer and keratoacanthoma in Filipinos: an incidence report from Kauai, Hawaii. Int. J. Dermatol. 1993;32:717–718. doi: 10.1111/j.1365-4362.1993.tb02740.x. [DOI] [PubMed] [Google Scholar]
- 9.Hoshida Y, et al. Cancer risk after renal transplantation in Japan. Int. J. Cancer. 1997;71:517–520. doi: 10.1002/(sici)1097-0215(19970516)71:4<517::aid-ijc3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 10.Kricker A, Armstrong BK, English DR, Heenan PJ. Does intermittent sun exposure cause basal cell carcinoma? A case-control study in Western Australia. Int. J. Cancer. 1995;60:489–494. doi: 10.1002/ijc.2910600411. [DOI] [PubMed] [Google Scholar]
- 11.Rosso S, et al. The multicentre south European study ‘Helios’. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br. J. Cancer. 1996;73:1447–1454. doi: 10.1038/bjc.1996.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson SC, Jolley D, Marks R. Reduction of solar keratoses by regular sunscreen use. N. Engl. J. Med. 1993;329:1147–1151. doi: 10.1056/NEJM199310143291602. [DOI] [PubMed] [Google Scholar]
- 13.Pandeya N, Purdie DM, Green A, Williams G. Repeated occurrence of basal cell carcinoma of the skin and multifailure survival analysis: follow-up data from the Nambour Skin Cancer Prevention Trial. Am. J. Epidemiol. 2005;161:748–754. doi: 10.1093/aje/kwi098. [DOI] [PubMed] [Google Scholar]
- 14.van der Pols JC, Williams GM, Pandeya N, Logan V, Green AC. Prolonged prevention of squamous cell carcinoma of the skin by regular sunscreen use. Cancer Epidemiol. Biomarkers Prev. 2006;15:2546–2548. doi: 10.1158/1055-9965.EPI-06-0352. [DOI] [PubMed] [Google Scholar]
- 15.Kricker A, Armstrong BK, English DR, Heenan PJ. A dose-response curve for sun exposure and basal cell carcinoma. Int. J. Cancer. 1995;60:482–488. doi: 10.1002/ijc.2910600410. [DOI] [PubMed] [Google Scholar]
- 16.Guo HR, Yu HS, Hu H, Monson RR. Arsenic in drinking water and skin cancers: cell-type specificity (Taiwan, ROC) Cancer Causes Control. 2001;12:909–916. doi: 10.1023/a:1013712203455. [DOI] [PubMed] [Google Scholar]
- 17.Karagas MR, et al. Skin cancer risk in relation to toenail arsenic concentrations in a US population- based case-control study. Am. J. Epidemiol. 2001;153:559–565. doi: 10.1093/aje/153.6.559. [DOI] [PubMed] [Google Scholar]
- 18.Karagas MR, Stukel TA, Tosteson TD. Assessment of cancer risk and environmental levels of arsenic in New Hampshire. Int. J. Hyg. Environ. Health. 2002;205:85–94. doi: 10.1078/1438-4639-00133. [DOI] [PubMed] [Google Scholar]
- 19.Gailani MR, et al. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell. 1992;69:111–117. doi: 10.1016/0092-8674(92)90122-s. [DOI] [PubMed] [Google Scholar]
- 20.Hahn H, et al. Mutations of the human homologue of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 21.Johnson RL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. References 19–21 provide the original data linking basal cell carcinogenesis to aberrant activation of HH signalling. [DOI] [PubMed] [Google Scholar]
- 22.Klein RD, Dykas DJ, Bale AE. Clinical testing for the nevoid basal cell carcinoma syndrome in a DNA diagnostic laboratory. Genet. Med. 2005;7:611–619. doi: 10.1097/01.gim.0000182879.57182.b4. [DOI] [PubMed] [Google Scholar]
- 23.Ling G, et al. PATCHED and p53 gene alterations in sporadic and hereditary basal cell cancer. Oncogene. 2001;20:7770–7778. doi: 10.1038/sj.onc.1204946. [DOI] [PubMed] [Google Scholar]
- 24.Ouhtit A, et al. UV-radiation-specific p53 mutation frequency in normal skin as a predictor of risk of basal cell carcinoma. J. Natl Cancer Inst. 1998;90:523–531. doi: 10.1093/jnci/90.7.523. [DOI] [PubMed] [Google Scholar]
- 25.Hutchin ME, et al. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005;19:214–223. doi: 10.1101/gad.1258705. This paper illustrates the requirement for continued HH signalling for BCC maintenance in a murine model of HH-driven BCC carcinogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adolphe C, Hetherington R, Ellis T, Wainwright B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006;66:2081–2088. doi: 10.1158/0008-5472.CAN-05-2146. [DOI] [PubMed] [Google Scholar]
- 27.Gailani MR, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nature Genet. 1996;14:78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 28.Aszterbaum M, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J. Invest. Dermatol. 1998;110:885–888. doi: 10.1046/j.1523-1747.1998.00222.x. [DOI] [PubMed] [Google Scholar]
- 29.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 30.Reifenberger J, et al. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998;58:1798–1803. [PubMed] [Google Scholar]
- 31.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–1759. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 32.Rohatgi R, Scott MP. Patching the gaps in Hedgehog signalling. Nature Cell Biol. 2007;9:1005–1009. doi: 10.1038/ncb435. [DOI] [PubMed] [Google Scholar]
- 33.Varjosalo M, Taipale J. Hedgehog signaling. J. Cell Sci. 2007;120:3–6. doi: 10.1242/jcs.03309. [DOI] [PubMed] [Google Scholar]
- 34.Kinzler KW, et al. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 35.Bhatia N, et al. Gli2 is targeted for ubiquitination and degradation by β-TrCP ubiquitin ligase. J. Biol. Chem. 2006;281:19320–19326. doi: 10.1074/jbc.M513203200. [DOI] [PubMed] [Google Scholar]
- 36.Huntzicker EG, et al. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006;20:276–281. doi: 10.1101/gad.1380906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle. 2006;5:2457–2463. doi: 10.4161/cc.5.21.3406. [DOI] [PubMed] [Google Scholar]
- 38.Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huangfu D, et al. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 40.Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl Acad. Sci. USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corbit KC, et al. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 42.May SR, et al. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 43.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 44.Bonifas JM, et al. Activation of expression of hedgehog target genes in basal cell carcinomas. J. Invest. Dermatol. 2001;116:739–742. doi: 10.1046/j.1523-1747.2001.01315.x. [DOI] [PubMed] [Google Scholar]
- 45.Tojo M, Kiyosawa H, Iwatsuki K, Kaneko F. Expression of a sonic hedgehog signal transducer, hedgehog-interacting protein, by human basal cell carcinoma. Br. J. Dermatol. 2002;146:69–73. doi: 10.1046/j.1365-2133.2002.04583.x. [DOI] [PubMed] [Google Scholar]
- 46.Ashton KJ, Weinstein SR, Maguire DJ, Griffiths LR. Molecular cytogenetic analysis of basal cell carcinoma DNA using comparative genomic hybridization. J. Invest. Dermatol. 2001;117:683–686. doi: 10.1046/j.0022-202x.2001.01434.x. [DOI] [PubMed] [Google Scholar]
- 47.Reifenberger J, et al. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005;152:43–51. doi: 10.1111/j.1365-2133.2005.06353.x. [DOI] [PubMed] [Google Scholar]
- 48.Lindstrom E, Shimokawa T, Toftgard R, Zaphiropoulos PG. PTCH mutations: distribution and analyses. Hum. Mutat. 2006;27:215–219. doi: 10.1002/humu.20296. [DOI] [PubMed] [Google Scholar]
- 49.Bodak N, et al. High levels of patched gene mutations in basal-cell carcinomas from patients with xeroderma pigmentosum. Proc. Natl Acad. Sci. USA. 1999;96:5117–5122. doi: 10.1073/pnas.96.9.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daya-Grosjean L, Sarasin A. UV-specific mutations of the human patched gene in basal cell carcinomas from normal individuals and xeroderma pigmentosum patients. Mutat. Res. 2000;450:193–199. doi: 10.1016/s0027-5107(00)00025-7. [DOI] [PubMed] [Google Scholar]
- 51.Couve-Privat S, Bouadjar B, Avril MF, Sarasin A, Daya-Grosjean L. Significantly high levels of ultraviolet-specific mutations in the smoothened gene in basal cell carcinomas from DNA repair-deficient xeroderma pigmentosum patients. Cancer Res. 2002;62:7186–7189. [PubMed] [Google Scholar]
- 52.Moriwaki S, Ray S, Tarone RE, Kraemer KH, Grossman L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: reduced DNA repair capacity and increased DNA mutability. Mutat. Res. 1996;364:117–123. doi: 10.1016/0921-8777(96)00029-8. [DOI] [PubMed] [Google Scholar]
- 53.Rees JL. The genetics of sun sensitivity in humans. Am. J. Hum. Genet. 2004;75:739–751. doi: 10.1086/425285. This remains an authoritative review of genetic factors predisposing to UV-induced skin cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han J, Kraft P, Colditz GA, Wong J, Hunter DJ. Melanocortin 1 receptor variants and skin cancer risk. Int. J. Cancer. 2006;119:1976–1984. doi: 10.1002/ijc.22074. [DOI] [PubMed] [Google Scholar]
- 55.Liboutet M, et al. MC1R and PTCH gene polymorphism in French patients with basal cell carcinomas. J. Invest. Dermatol. 2006;126:1510–1517. doi: 10.1038/sj.jid.5700263. [DOI] [PubMed] [Google Scholar]
- 56.Box NF, et al. Melanocortin-1 receptor genotype is a risk factor for basal and squamous cell carcinoma. J. Invest. Dermatol. 2001;116:224–229. doi: 10.1046/j.1523-1747.2001.01224.x. [DOI] [PubMed] [Google Scholar]
- 57.Bastiaens MT, et al. Melanocortin-1 receptor gene variants determine the risk of nonmelanoma skin cancer independently of fair skin and red hair. Am. J. Hum. Genet. 2001;68:884–894. doi: 10.1086/319500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerstenblith MR, Goldstein AM, Fargnoli MC, Peris K, Landi MT. Comprehensive evaluation of allele frequency differences of MC1R variants across populations. Hum. Mutat. 2007;28:495–505. doi: 10.1002/humu.20476. [DOI] [PubMed] [Google Scholar]
- 59.Palmer JS, et al. Melanocortin-1 receptor polymorphisms and risk of melanoma: is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet. 2000;66:176–186. doi: 10.1086/302711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kennedy C, et al. Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J. Invest. Dermatol. 2001;117:294–300. doi: 10.1046/j.0022-202x.2001.01421.x. [DOI] [PubMed] [Google Scholar]
- 61.Slominski A, Paus R, Wortsman J. Can some melanotropins modulate keratinocyte proliferation? J. Invest. Dermatol. 1991;97, 747 doi: 10.1111/1523-1747.ep12484829. [DOI] [PubMed] [Google Scholar]
- 62.Wintzen M, Yaar M, Burbach JP, Gilchrest BA. Proopiomelanocortin gene product regulation in keratinocytes. J. Invest. Dermatol. 1996;106:673–678. doi: 10.1111/1523-1747.ep12345496. [DOI] [PubMed] [Google Scholar]
- 63.Corre S, et al. UV-induced expression of key component of the tanning process, the POMC and MC1R genes, is dependent on the p-38-activated upstream stimulating factor-1 (USF-1) J. Biol. Chem. 2004;279:51226–51233. doi: 10.1074/jbc.M409768200. [DOI] [PubMed] [Google Scholar]
- 64.Cui R, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128:853–864. doi: 10.1016/j.cell.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 65.Gudbjartsson DF, et al. ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nature Genet. 2008;40:886–891. doi: 10.1038/ng.161. This is a recent large-scale survey of genetic variants predisposing to BCC carcinogenesis. [DOI] [PubMed] [Google Scholar]
- 66.Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. Arch. Dermatol. 1994;130:1018–1021. [PubMed] [Google Scholar]
- 67.Berwick M, Vineis P. Markers of DNA repair and susceptibility to cancer in humans: an epidemiologic review. J. Natl Cancer Inst. 2000;92:874–897. doi: 10.1093/jnci/92.11.874. [DOI] [PubMed] [Google Scholar]
- 68.Wei Q, Matanoski GM, Farmer ER, Hedayati MA, Grossman L. DNA repair capacity for ultraviolet light-induced damage is reduced in peripheral lymphocytes from patients with basal cell carcinoma. J. Invest. Dermatol. 1995;104:933–936. doi: 10.1111/1523-1747.ep12606207. [DOI] [PubMed] [Google Scholar]
- 69.Dybdahl M, Frentz G, Vogel U, Wallin H, Nexo BA. Low DNA repair is a risk factor in skin carcinogenesis: a study of basal cell carcinoma in psoriasis patients. Mutat. Res. 1999;433:15–22. doi: 10.1016/s0921-8777(98)00057-3. [DOI] [PubMed] [Google Scholar]
- 70.Segerback D, Strozyk M, Snellman E, Hemminki K. Repair of UV dimers in skin DNA of patients with basal cell carcinoma. Cancer Epidemiol. Biomarkers Prev. 2008;17:2388–2392. doi: 10.1158/1055-9965.EPI-08-0248. [DOI] [PubMed] [Google Scholar]
- 71.Han S, et al. DNA repair gene XRCC3 polymorphisms and cancer risk: a meta-analysis of 48 case-control studies. Eur. J. Hum. Genet. 2006;14:1136–1144. doi: 10.1038/sj.ejhg.5201681. [DOI] [PubMed] [Google Scholar]
- 72.Han J, Colditz GA, Samson LD, Hunter DJ. Polymorphisms in DNA double-strand break repair genes and skin cancer risk. Cancer Res. 2004;64:3009–3013. doi: 10.1158/0008-5472.can-04-0246. [DOI] [PubMed] [Google Scholar]
- 73.Thirumaran RK, et al. Single nucleotide polymorphisms in DNA repair genes and basal cell carcinoma of skin. Carcinogenesis. 2006;27:1676–1681. doi: 10.1093/carcin/bgi381. [DOI] [PubMed] [Google Scholar]
- 74.Jacobsen NR, et al. No association between the DNA repair gene XRCC3 T241M polymorphism and risk of skin cancer and breast cancer. Cancer Epidemiol. Biomarkers Prev. 2003;12:584–585. [PubMed] [Google Scholar]
- 75.Festa F, et al. Basal cell carcinoma and variants in genes coding for immune response, DNA repair, folate and iron metabolism. Mutat. Res. 2005;574:105–111. doi: 10.1016/j.mrfmmm.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 76.Chen YC, et al. Genetic polymorphism in p53 codon 72 and skin cancer in southwestern Taiwan. J. Environ. Sci. Health A Tox Hazard Subst. Environ. Eng. 2003;38:201–211. doi: 10.1081/ese-120016889. [DOI] [PubMed] [Google Scholar]
- 77.Han J, Cox DG, Colditz GA, Hunter DJ. The p53 codon 72 polymorphism, sunburns, and risk of skin cancer in US Caucasian women. Mol. Carcinog. 2006;45:694–700. doi: 10.1002/mc.20190. [DOI] [PubMed] [Google Scholar]
- 78.Stefanaki I, et al. p53 codon 72 Pro homozygosity increases the risk of cutaneous melanoma in individuals with dark skin complexion and among noncarriers of melanocortin 1 receptor red hair variants. Br. J. Dermatol. 2007;156:357–362. doi: 10.1111/j.1365-2133.2006.07645.x. [DOI] [PubMed] [Google Scholar]
- 79.McGregor JM, et al. Relationship between p53 codon 72 polymorphism and susceptibility to sunburn and skin cancer. J. Invest. Dermatol. 2002;119:84–90. doi: 10.1046/j.1523-1747.2002.01655.x. [DOI] [PubMed] [Google Scholar]
- 80.Wilkening S, et al. No association between MDM2 SNP309 promoter polymorphism and basal cell carcinoma of the skin. Br. J. Dermatol. 2007;157:375–377. doi: 10.1111/j.1365-2133.2007.07994.x. [DOI] [PubMed] [Google Scholar]
- 81.Asplund A, et al. PTCH codon 1315 polymorphism and risk for nonmelanoma skin cancer. Br. J. Dermatol. 2005;152:868–873. doi: 10.1111/j.1365-2133.2005.06464.x. [DOI] [PubMed] [Google Scholar]
- 82.Wakabayashi Y, Mao JH, Brown K, Girardi M, Balmain A. Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature. 2007;445:761–765. doi: 10.1038/nature05489. [DOI] [PubMed] [Google Scholar]
- 83.Yoon JW, et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1- induced cell transformation. J. Biol. Chem. 2002;277:5548–5555. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- 84.Howell BG, et al. Microarray profiles of human basal cell carcinoma: insights into tumor growth and behavior. J. Dermatol. Sci. 2005;39:39–51. doi: 10.1016/j.jdermsci.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 85.O’Driscoll L, et al. Investigation of the molecular profile of basal cell carcinoma using whole genome microarrays. Mol. Cancer. 2006;5, 74 doi: 10.1186/1476-4598-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu M, et al. Superficial, nodular, and morpheiform basal-cell carcinomas exhibit distinct gene expression profiles. J. Invest. Dermatol. 2008;128:1797–1805. doi: 10.1038/sj.jid.5701243. [DOI] [PubMed] [Google Scholar]
- 87.Asplund A, et al. Expression profiling of microdissected cell populations selected from basal cells in normal epidermis and basal cell carcinoma. Br. J. Dermatol. 2008;158:527–538. doi: 10.1111/j.1365-2133.2007.08418.x. [DOI] [PubMed] [Google Scholar]
- 88.Xie J, et al. A role of PDGFRα in basal cell carcinoma proliferation. Proc. Natl Acad. Sci. USA. 2001;98:9255–9259. doi: 10.1073/pnas.151173398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bigelow RL, et al. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J. Biol. Chem. 2004;279:1197–1205. doi: 10.1074/jbc.M310589200. [DOI] [PubMed] [Google Scholar]
- 90.Regl G, et al. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004;64:7724–7731. doi: 10.1158/0008-5472.CAN-04-1085. [DOI] [PubMed] [Google Scholar]
- 91.Kump E, Ji J, Wernli M, Hausermann P, Erb P. Gli2 upregulates cFlip and renders basal cell carcinoma cells resistant to death ligand-mediated apoptosis. Oncogene. 2008;27:3856–3864. doi: 10.1038/onc.2008.5. [DOI] [PubMed] [Google Scholar]
- 92.Li C, et al. IFNα induces Fas expression and apoptosis in hedgehog pathway activated BCC cells through inhibiting Ras–Erk signaling. Oncogene. 2004;23:1608–1617. doi: 10.1038/sj.onc.1207273. [DOI] [PubMed] [Google Scholar]
- 93.Athar M, et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004;64:7545–7552. doi: 10.1158/0008-5472.CAN-04-1393. [DOI] [PubMed] [Google Scholar]
- 94.Leung C, et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 95.Reinisch CM, Uthman A, Erovic BM, Pammer J. Expression of BMI-1 in normal skin and inflammatory and neoplastic skin lesions. J. Cutan. Pathol. 2007;34:174–180. doi: 10.1111/j.1600-0560.2006.00587.x. [DOI] [PubMed] [Google Scholar]
- 96.Kenney AM, Rowitch DH. Sonic hedgehog promotes G cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol. Cell. Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rao G, et al. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23:6156–6162. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 98.Hahn H, et al. Patched target Igf2 is indispensable for the formation of medulloblastoma and rhabdomyosarcoma. J. Biol. Chem. 2000;15:28341–28344. doi: 10.1074/jbc.C000352200. [DOI] [PubMed] [Google Scholar]
- 99.Levitt RJ, Zhao Y, Blouin MJ, Pollak M. The hedgehog pathway inhibitor cyclopamine increases levels of p27, and decreases both expression of IGF-II and activation of Akt in PC-3 prostate cancer cells. Cancer Lett. 2007;255:300–306. doi: 10.1016/j.canlet.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 100.Lipinski RJ, et al. Sonic hedgehog signaling regulates the expression of insulin-like growth factor binding protein-6 during fetal prostate development. Dev. Dyn. 2005;233:829–836. doi: 10.1002/dvdy.20414. [DOI] [PubMed] [Google Scholar]
- 101.Allan GJ, et al. Major components of the insulin-like growth factor axis are expressed early in chicken embryogenesis, with IGF binding protein (IGFBP)-5 expression subject to regulation by sonic hedgehog. Anat. Embryol. (Berl.) 2003;207:73–84. doi: 10.1007/s00429-003-0321-x. [DOI] [PubMed] [Google Scholar]
- 102.Elia D, Madhala D, Ardon E, Reshef R, Halevy O. Sonic hedgehog promotes proliferation and differentiation of adult muscle cells: involvement of MAPK/ERK and PI3K/Akt pathways. Biochim. Biophys. Acta. 2007;1773:1438–1446. doi: 10.1016/j.bbamcr.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 103.Riobo NA, Lu K, Ai X, Haines GM, Emerson CP., Jr. Phosphoinositide 3-kinase and Akt are essential for sonic hedgehog signaling. Proc. Natl Acad. Sci. USA. 2006;103:4505–4510. doi: 10.1073/pnas.0504337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Teh MT, et al. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62:4773–4780. [PubMed] [Google Scholar]
- 105.Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007;132:1420–1431. doi: 10.1053/j.gastro.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 106.Dai B, et al. Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene. 2007;26:6212–6219. doi: 10.1038/sj.onc.1210443. [DOI] [PubMed] [Google Scholar]
- 107.Liu M, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–3602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- 108.Kalin TV, et al. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006;66:1712–1720. doi: 10.1158/0008-5472.CAN-05-3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim IM, et al. The forkhead box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–2161. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 110.Tan Y, Raychaudhuri P, Costa RH. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell. Biol. 2007;27:1007–1016. doi: 10.1128/MCB.01068-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schuller U, et al. Forkhead transcription factor FoxM1 regulates mitotic entry and prevents spindle defects in cerebellar granule neuron precursors. Mol. Cell. Biol. 2007;27:8259–8270. doi: 10.1128/MCB.00707-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wonsey DR, Follettie MT. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005;65:5181–5189. doi: 10.1158/0008-5472.CAN-04-4059. [DOI] [PubMed] [Google Scholar]
- 113.Kalinichenko VV, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–850. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gusarova GA, et al. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J. Clin. Invest. 2007;117:99–111. doi: 10.1172/JCI27527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Radhakrishnan SK, et al. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006;66:9731–9735. doi: 10.1158/0008-5472.CAN-06-1576. [DOI] [PubMed] [Google Scholar]
- 116.Eichberger T, et al. FOXE1, a new transcriptional target of GLI2, is expressed in human epidermis and basal cell carcinoma. J. Invest. Dermatol. 2004;122:1180–1187. doi: 10.1111/j.0022-202X.2004.22505.x. [DOI] [PubMed] [Google Scholar]
- 117.Brancaccio A, et al. Requirement of the forkhead gene Foxe1, a target of sonic hedgehog signaling, in hair follicle morphogenesis. Hum. Mol. Genet. 2004;13:2595–2606. doi: 10.1093/hmg/ddh292. [DOI] [PubMed] [Google Scholar]
- 118.Yang SH, et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β-catenin signaling. Nature Genet. 2008;40:1130–1135. doi: 10.1038/ng.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nilsson M, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl Acad. Sci. USA. 2000;97:3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grachtchouk M, et al. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nature Genet. 2000;24:216–217. doi: 10.1038/73417. [DOI] [PubMed] [Google Scholar]
- 121.Aszterbaum M, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nature Med. 1999;5:1285–1291. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- 122.Svard J, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 123.Grachtchouk V, et al. The magnitude of hedgehog signaling activity defines skin tumor phenotype. EMBO J. 2003;22:2741–2751. doi: 10.1093/emboj/cdg271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61:513–516. [PubMed] [Google Scholar]
- 125.Hadley ME, Dorr RT. Melanocortin peptide therapeutics: historical milestones, clinical studies and commercialization. Peptides. 2006;27:921–930. doi: 10.1016/j.peptides.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 126.Gange RW, Blackett AD, Matzinger EA, Sutherland BM, Kochevar IE. Comparative protection efficiency of UVA- and UVB-induced tans against erythema and formation of endonuclease sensitive sites in DNA by UVB in human skin. J. Invest. Dermatol. 1985;85:362–364. doi: 10.1111/1523-1747.ep12276983. [DOI] [PubMed] [Google Scholar]
- 127.Eller MS, Asarch A, Gilchrest BA. Photoprotection in human skin — a multifaceted SOS response. Photochem. Photobiol. 2008;84:339–349. doi: 10.1111/j.1751-1097.2007.00264.x. [DOI] [PubMed] [Google Scholar]
- 128.Arad S, et al. Topical thymidine dinucleotide treatment reduces development of ultraviolet-induced basal cell carcinoma in Ptch-1+/− mice. Am. J. Pathol. 2008;172:1248–1255. doi: 10.2353/ajpath.2008.071117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yarosh DB, Klein J. DNA repair enzymes in prevention of photocarcinogenesis. Photochem. Photobiol. 1996;63:445–447. doi: 10.1111/j.1751-1097.1996.tb03067.x. [DOI] [PubMed] [Google Scholar]
- 130.Yarosh D, et al. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet. 2001;357:926–929. doi: 10.1016/s0140-6736(00)04214-8. [DOI] [PubMed] [Google Scholar]
- 131.Peck GL, et al. Treatment and prevention of basal cell carcinoma with oral isotretinoin. J. Am. Acad. Dermatol. 1988;19:176–185. doi: 10.1016/s0190-9622(88)70162-0. [DOI] [PubMed] [Google Scholar]
- 132.Goldberg LH, Hsu SH, Alcalay J. Effectiveness of isotretinoin in preventing the appearance of basal cell carcinomas in basal cell nevus syndrome. J. Am. Acad. Dermatol. 1989;21:144–145. doi: 10.1016/s0190-9622(89)80359-7. [DOI] [PubMed] [Google Scholar]
- 133.Kraemer KH, DiGiovanna JJ, Moshell AN, Tarone RE, Peck GL. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N. Engl. J. Med. 1988;318:1633–1637. doi: 10.1056/NEJM198806233182501. [DOI] [PubMed] [Google Scholar]
- 134.Tangrea JA, et al. Long-term therapy with low-dose isotretinoin for prevention of basal cell carcinoma: a multicenter clinical trial. J. Natl Cancer Inst. 1992;84:328–332. doi: 10.1093/jnci/84.5.328. [DOI] [PubMed] [Google Scholar]
- 135.Levine N, et al. Trial of retinol and isotretinoin in skin cancer prevention: a randomized double-blind, controlled trial. Cancer Epidemiol. Biomarkers Prevention. 1997;6:957–961. [PubMed] [Google Scholar]
- 136.Peris K, Fargnoli MC, Chimenti S. Preliminary observations on the use of topical tazarotene to treat basal-cell carcinoma. N. Engl. J. Med. 1999;341:1767–1768. doi: 10.1056/NEJM199912023412312. [DOI] [PubMed] [Google Scholar]
- 137.Duvic M, et al. Tazarotene-induced gene 3 is suppressed in basal cell carcinomas and reversed in vivo by tazarotene application. J. Invest. Dermatol. 2003;121:902–909. doi: 10.1046/j.1523-1747.2003.12488.x. [DOI] [PubMed] [Google Scholar]
- 138.Bianchi L, et al. Topical treatment of basal cell carcinoma with tazarotene: a clinicopathological study on a large series of cases. Br. J. Dermatol. 2004;151:148–156. doi: 10.1111/j.1365-2133.2004.06044.x. [DOI] [PubMed] [Google Scholar]
- 139.So PL, et al. Topical tazarotene chemoprevention reduces basal cell carcinoma number and size in Ptch1+/− mice exposed to ultraviolet or ionizing radiation. Cancer Res. 2004;64:4385–4389. doi: 10.1158/0008-5472.CAN-03-1927. [DOI] [PubMed] [Google Scholar]
- 140.So PL, Fujimoto MA, Epstein EH., Jr Pharmacologic retinoid signaling and physiologic retinoic acid receptor signaling inhibit basal cell carcinoma tumorigenesis. Mol. Cancer Ther. 2008;7:1275–1284. doi: 10.1158/1535-7163.MCT-07-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Black HS, et al. Evidence that a low-fat diet reduces the occurence of non-melanoma skin cancer. Int. J. Cancer. 1995;62:165–169. doi: 10.1002/ijc.2910620210. [DOI] [PubMed] [Google Scholar]
- 142.Carneiro BA, Watkin WG, Mehta UK, Brockstein BE. Metastatic basal cell carcinoma: complete response to chemotherapy and associated pure red cell aplasia. Cancer Invest. 2006;24:396–400. doi: 10.1080/07357900600705474. [DOI] [PubMed] [Google Scholar]
- 143.Binns W, James LF, Shupe JL, Everett G. A congenital cyclopian-type malformation in lambs induced by maternal ingestion of a range plant, Veratrum californicum. Am. J. Vet. Res. 1963;24:1164–1175. [PubMed] [Google Scholar]
- 144.Tabs S, Avci O. Induction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivity. Eur. J. Dermatol. 2004;14:96–102. [PubMed] [Google Scholar]
- 145.Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 146.Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development. 1998;125:3553–3562. doi: 10.1242/dev.125.18.3553. [DOI] [PubMed] [Google Scholar]
- 147.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Taipale J, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 149.Van Hoff DD, et al. Efficacy data of GDC-0449, a systemic Hedgehog (Hh) pathway antagonist, in a first-in-human, first-in-class, phase I study with locally advanced, multifocal or metastatic basal cell carcinoma patients. Proc. 99th Annu. Meeting Am. Assoc. Cancer Res. abstract LB. 2008;138 [Google Scholar]
- 150.Palma V, et al. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132:335–344. doi: 10.1242/dev.01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Balordi F, Fishell G. Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J. Neurosci. 2007;27:14248–14259. doi: 10.1523/JNEUROSCI.4531-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang CL, Zou Y, He W, Gage FH, Evans RM. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature. 2008;451:1004–1007. doi: 10.1038/nature06562. [DOI] [PubMed] [Google Scholar]
- 153.Paladini RD, Saleh J, Qian C, Xu GX, Rubin LL. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J. Invest. Dermatol. 2005;125:638–646. doi: 10.1111/j.0022-202X.2005.23867.x. [DOI] [PubMed] [Google Scholar]
- 154.Miura H, Kusakabe Y, Harada S. Cell lineage and differentiation in taste buds. Arch. Histol. Cytol. 2006;69:209–225. doi: 10.1679/aohc.69.209. [DOI] [PubMed] [Google Scholar]
- 155.Angot E, et al. Chemoattractive activity of sonic hedgehog in the adult subventricular zone modulates the number of neural precursors reaching the olfactory bulb. Stem Cells. 2008 Jul;10 doi: 10.1634/stemcells.2008-0297. [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 156.Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13:249–260. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 157.Arad S, Konnikov N, Goukassian DA, Gilchrest BA. Quantification of inducible SOS-like photoprotective responses in human skin. J. Invest. Dermatol. 2007;127:2629–2636. doi: 10.1038/sj.jid.5700893. [DOI] [PubMed] [Google Scholar]
- 158.Bijlsma MF, et al. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol. 2006;4, e232 doi: 10.1371/journal.pbio.0040232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Holick MF, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. 1980;210:203–205. doi: 10.1126/science.6251551. [DOI] [PubMed] [Google Scholar]
- 160.MacLaughlin J, Holick MF. Aging decreases the capacity of human skin to produce vitamin D3. J. Clin. Invest. 1985;76:1536–1538. doi: 10.1172/JCI112134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vogt A, Hebert J, Hwang J, Lu Y, Epstein EH. Anti-rejection drug treatment increases basal cell carcinoma burden in Ptch1+/− mice. J. Invest. Dermatol. 2005;124:263–267. doi: 10.1111/j.0022-202X.2004.23573.x. [DOI] [PubMed] [Google Scholar]
- 162.Karhadkar SS, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 163.Scott EJ, Reinertson RP. The modulating influence of stromal environment on epithelial cells studies in human autoransplants. J. Invest. Dermatol. 1961;36:109–131. [PubMed] [Google Scholar]
- 164.Williams T, et al. The oncogenic GLI (GLI1 and GLI2) transcription factors induce characteristics of cellular senescence in N/Tert-1 keratinocytes. J. Invest. Dermatol. 2007;127, s92 [Google Scholar]
- 165.Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987;66:98–113. doi: 10.1097/00005792-198703000-00002. [DOI] [PubMed] [Google Scholar]
- 166.Evans DGR, Farndon PA, Burnell LD, Gattamaneni HR, Birch JM. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br. J. Cancer. 1991;64:959–961. doi: 10.1038/bjc.1991.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Johnson AD, Hebert AA, Esterly NB. Nevoid basal cell carcinoma syndrome: bilateral ovarian fibromas in a 3 1/2-year-old girl. J. Am. Acad. Dermatol. 1986;14:371–374. doi: 10.1016/s0190-9622(86)70046-7. References 165–167 review the clinical aspects of BCNS. [DOI] [PubMed] [Google Scholar]
- 168.Kimonis VE, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am. J. Med. Genet. 1997;69:299–308. [PubMed] [Google Scholar]
- 169.Miller KL, et al. XPA, haplotypes, and risk of basal and squamous cell carcinoma. Carcinogenesis. 2006;27:1670–1675. doi: 10.1093/carcin/bgi376. [DOI] [PubMed] [Google Scholar]
- 170.Kang SY, et al. Polymorphisms in the DNA repair gene XRCC1 associated with basal cell carcinoma and squamous cell carcinoma of the skin in a Korean population. Cancer Sci. 2007;98:716–720. doi: 10.1111/j.1349-7006.2007.00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Han J, Hankinson SE, Colditz GA, Hunter DJ. Genetic variation in XRCC1, sun exposure, and risk of skin cancer. Br. J. Cancer. 2004;91:1604–1609. doi: 10.1038/sj.bjc.6602174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dybdahl M, Vogel U, Frentz G, Wallin H, Nexo BA. Polymorphisms in the DNA repair gene XPD: correlations with risk and age at onset of basal cell carcinoma. Cancer Epidemiol. Biomarkers Prev. 1999;8:77–81. [PubMed] [Google Scholar]
- 173.Lovatt T, et al. Polymorphism in the nuclear excision repair gene ERCC2/XPD: association between an exon 6–exon 10 haplotype and susceptibility to cutaneous basal cell carcinoma. Hum. Mutat. 2005;25:353–359. doi: 10.1002/humu.20158. [DOI] [PubMed] [Google Scholar]
- 174.Vogel U, et al. Effect of polymorphisms in XPD, RAI, ASE-1 and ERCC1 on the risk of basal cell carcinoma among Caucasians after age 50. Cancer Detect. Prev. 2005;29:209–214. doi: 10.1016/j.cdp.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 175.Han J, Colditz GA, Hunter DJ. Lack of associations of selected variants in genes involved in cell cycle and apoptosis with skin cancer risk. Cancer Epidemiol. Biomarkers Prev. 2006;15:592–593. doi: 10.1158/1055-9965.EPI-05-0915. [DOI] [PubMed] [Google Scholar]
- 176.Lin Q, et al. Increased susceptibility to UV-induced skin carcinogenesis in polymerase eta-deficient mice. Cancer Res. 2006;66:87–94. doi: 10.1158/0008-5472.CAN-05-1862. [DOI] [PubMed] [Google Scholar]
- 177.Meeran SM, Mantena SK, Meleth S, Elmets CA, Katiyar SK. Interleukin-12-deficient mice are at greater risk of UV radiation-induced skin tumors and malignant transformation of papillomas to carcinomas. Mol. Cancer Ther. 2006;5:825–832. doi: 10.1158/1535-7163.MCT-06-0003. [DOI] [PubMed] [Google Scholar]
- 178.Schwarz T. Photoimmunosuppression. Photodermatol. Photoimmunol. Photomed. 2002;18:141–145. doi: 10.1034/j.1600-0781.2002.180307.x. [DOI] [PubMed] [Google Scholar]
- 179.Reynolds NJ, Todd C, Angus B. Overexpression of protein kinase C-α and -β isozymes by stromal dendritic cells in basal and squamous cell carcinoma. Br. J. Dermatol. 1997;136:666–673. [PubMed] [Google Scholar]
- 180.Neill GW, et al. Loss of protein kinase Cα expression may enhance the tumorigenic potential of Gli1 in basal cell carcinoma. Cancer Res. 2003;63:4692–4697. [PubMed] [Google Scholar]
- 181.Riobo NA, Haines GM, Emerson CP., Jr Protein kinase C-δ and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006;66:839–845. doi: 10.1158/0008-5472.CAN-05-2539. [DOI] [PubMed] [Google Scholar]
- 182.Lauth M, Bergstrom A, Toftgard R. Phorbol esters inhibit the Hedgehog signalling pathway downstream of Suppressor of Fused, but upstream of Gli. Oncogene. 2007;26:5163–5168. doi: 10.1038/sj.onc.1210321. [DOI] [PubMed] [Google Scholar]
- 183.Tang X, et al. Ornithine decarboxylase is a target for chemoprevention of basal and squamous cell carcinomas in Ptch1+/− mice. J. Clin. Invest. 2004;113:867–875. doi: 10.1172/JCI20732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thyberg J, Fredholm BB. Induction of ornithine decarboxylase activity and putrescine synthesis in arterial smooth muscle cells stimulated with platelet- derived growth factor. Exp. Cell Res. 1987;170:160–169. doi: 10.1016/0014-4827(87)90125-x. [DOI] [PubMed] [Google Scholar]
- 185.Kasper M, et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 2006;26:6283–6298. doi: 10.1128/MCB.02317-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mimeault M, et al. Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Mol. Cancer Ther. 2007;6:967–978. doi: 10.1158/1535-7163.MCT-06-0648. [DOI] [PubMed] [Google Scholar]
- 187.Ikeuchi T, Urano Y, Fukuhara K, Nakanishi H, Arase S. Light microscopic autoradiographical analysis of [125I]epidermal growth factor binding in basal cell epithelioma and squamous cell carcinoma of the skin. J. Dermatol. 1993;20:219–225. doi: 10.1111/j.1346-8138.1993.tb03865.x. [DOI] [PubMed] [Google Scholar]
- 188.Mimeault M, et al. Cytotoxic effects induced by a combination of cyclopamine and gefitinib, the selective hedgehog and epidermal growth factor receptor signaling inhibitors, in prostate cancer cells. Int. J. Cancer. 2006;118:1022–1031. doi: 10.1002/ijc.21440. [DOI] [PubMed] [Google Scholar]
- 189.Hu WG, Liu T, Xiong JX, Wang CY. Blockade of sonic hedgehog signal pathway enhances antiproliferative effect of EGFR inhibitor in pancreatic cancer cells. Acta Pharmacol. Sin. 2007;28:1224–1230. doi: 10.1111/j.1745-7254.2007.00620.x. [DOI] [PubMed] [Google Scholar]
- 190.Neill GW, et al. GLI1 repression of ERK activity correlates with colony formation and impaired migration in human epidermal keratinocytes. Carcinogenesis. 2008;29:738–746. doi: 10.1093/carcin/bgn037. [DOI] [PubMed] [Google Scholar]
- 191.Dennler S, et al. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67:6981–6986. doi: 10.1158/0008-5472.CAN-07-0491. [DOI] [PubMed] [Google Scholar]
- 192.Stamp GW, et al. Transforming growth factor-β distribution in basal cell carcinomas: relationship to proliferation index. Br. J. Dermatol. 1993;129:57–64. doi: 10.1111/j.1365-2133.1993.tb03312.x. [DOI] [PubMed] [Google Scholar]
- 193.Gambichler T, et al. Increased expression of TGF-β/Smad proteins in basal cell carcinoma. Eur. J. Med. Res. 2007;12:509–514. [PubMed] [Google Scholar]
- 194.Verhaegh ME, Arends JW, Majoie IM, Hoekzema R, Neumann HA. Transforming growth factor-β and bcl-2 distribution patterns distinguish trichoepithelioma from basal cell carcinoma. Dermatol. Surg. 1997;23:695–700. doi: 10.1111/j.1524-4725.1997.tb00392.x. [DOI] [PubMed] [Google Scholar]
- 195.Lange D, et al. Expression of TGF-β related Smad proteins in human epithelial skin tumors. Int. J. Oncol. 1999;14:1049–1056. doi: 10.3892/ijo.14.6.1049. [DOI] [PubMed] [Google Scholar]
- 196.Sneddon JB, et al. Bone morphogenetic protein antagonist gremlin 1 is widely expressed by cancer-associated stromal cells and can promote tumor cell proliferation. Proc. Natl Acad. Sci. USA. 2006;103:14842–14847. doi: 10.1073/pnas.0606857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Thelu J, Rossio P, Favier B. Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma, psoriasis and wound healing. BMC Dermatol. 2002;2, 7 doi: 10.1186/1471-5945-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Nicolas M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nature Genet. 2003;33:416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 199.Christenson LJ, et al. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA. 2005;294:681–690. doi: 10.1001/jama.294.6.681. [DOI] [PubMed] [Google Scholar]