

Abstract
The Drug Design Data Resource (D3R) ran Grand Challenge 2015 between September 2015 and February 2016. Two targets served as the framework to test community docking and scoring methods: (i) HSP90, donated by AbbVie and the Community Structure Activity Resource (CSAR), and (ii) MAP4K4, donated by Genentech. The challenges for both target datasets were conducted in two stages, with the first stage testing pose predictions and the capacity to rank compounds by affinity with minimal structural data; and the second stage testing methods for ranking compounds with knowledge of at least a subset of the ligand-protein poses. An additional sub-challenge provided small groups of chemically similar HSP90 compounds amenable to alchemical calculations of relative binding free energy. Unlike previous blinded Challenges, we did not provide cognate receptors or receptors prepared with hydrogens and likewise did not require a specified crystal structure to be used for pose or affinity prediction in Stage 1. Given the freedom to select from over 200 crystal structures of HSP90 in the PDB, participants employed workflows that tested not only core docking and scoring technologies, but also methods for addressing water-mediated ligand-protein interactions, binding pocket flexibility, and the optimal selection of protein structures for use in docking calculations. Nearly 40 participating groups submitted over 350 prediction sets for Grand Challenge 2015. This overview describes the datasets and the organization of the challenge components, summarizes the results across all submitted predictions, and considers broad conclusions that may be drawn from this collaborative community endeavor.
Keywords: D3R, docking, FEP, alchemical methods, ligand ranking, water mediated ligand interaction
1 Introduction
The Drug Design Data Resource (D3R; www.drugdesigndata.org), aims to motivate the development of improved computational methods by offering pharmaceutical-related high quality datasets as benchmarks and blinded community challenges. In this article we describe the composition of the datasets, the challenge preparation, and motivation for the challenge provided, and an overview of participant results of the first community-wide D3R Grand Challenge, conducted between September 15, 2015 and February 1, 2016. Grand Challenge 2015 had high participation and the approaches to solving this protein-ligand pose and affinity ranking problem-set were varied. This overview is supplemented in detail by a number of participant authored articles in this special issue.
Development of accurate automated protein-ligand docking and scoring methods promises a high social impact by reducing the time, expense, and environmental costs of extensive chemistry campaigns in drug discovery. However, this high potential is matched by the high barriers to developing such methods. Design goals include the abilities to recapitulate experimentally observed ligand-protein poses with root-mean-square deviations (RMSD) ≤ 2.0Å, and to accurately rank affinities for ligands bound to any receptor. At least three types of affinity ranking problems are commonly addressed: ranking of diverse compounds to classify them as binders or non-binders of a given protein (virtual screening); ranking of congeneric series of active compounds that bind a single protein; and ranking of the affinities of a single ligand against various proteins, particularly within a protein family. A number of community challenges in recent years have tested the performance of these algorithms, identifying successes and illuminating areas for improvement [1–4].
D3R’s Grand Challenge 2015 presented the community with two datasets from drug discovery programs at two different companies, Abbvie (formerly Abbott) and Genentech, Inc. The first sub-challenge focuses on the human heat shock protein 90 (HSP90), which regulates proteostasis in normal cells and the stabilization of many oncoproteins in tumor cells [5, 6]. Over the past fifteen years, discovery of small molecule inhibitors of the HSP90 ATP-binding domain has led to over 20 anticancer agents that are currently in clinical trials [6]. The second sub-challenge focused on the human mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4), a signaling Ser/Thr kinase that is involved in pathways regulating various pathological processes, including inflammation and cancer, and is therefore an investigational drug target for a range of diseases [7].
The HSP90 dataset is based on data contributed by Abbvie and was enhanced by our predecessor the Community Structure-Affinity Resource (CSAR www.csardock.org), which added 17 new compounds and related assay and structural data. This dataset exemplifies many of the features of an ideal docking challenge dataset representative of a drug discovery program. The dataset consists of three different chemical series of ~ 60 compounds each, which cover an affinity range of four orders of magnitude. Participants were provided with examples of chemically similar compounds that do not inhibit the target, as well as a set of co-crystal structures representative of the binding modes each chemical series. This challenge provided participants with the opportunity to test workflows that go beyond the docking algorithms and scoring methodologies and to find ways to incorporate the extensive knowledge base that exists for this target, including over 200 crystal structures in the Protein Data Bank (PDB; www.pdb.org) with various bound ligands, binding modes and conformational examples for most prevalent conformations of the binding pocket’s ATP-lid [8]. The distribution of affinities and the number of compounds provided in this dataset also afforded the opportunity to define subsets of chemically similar compounds designed to test alchemical methods [9] of computing relative binding free energies.
The second dataset is based on the protein MAP4K4, and was contributed by Genentech. This dataset contained 30 crystal structures with resolution limits better than 2.5 Å, and inhibition/binding data for 18 compounds spanning four orders of magnitude, in many cases confirmed by multiple assay methods. This dataset differs from HSP90 in that it has a high number of unrelated diverse compounds and only a third of the compounds fall into a congeneric series. Moreover, there was a paucity of co-crystal structures publicly available at the time of the Challenge launch; only eight ligand-bound (non-ATP) structures were extant in the PDB [7, 10–12]. Thus most of the compounds in the dataset were unrelated to available MAP4K4 crystal structures and provided an opportunity to test docking programs’ cross-docking ability. In MAP4K4, as in the HSP90 dataset, binding site flexibility adds to the difficulty of pose prediction, as the kinase P-loop can adopt both a closed and open conformation [13], and in some cases, has residues unresolved in crystal structures due to its flexibility [13].
Unlike previous blinded challenges, we chose to present these subchallenges without providing cognate receptors or receptors prepared with hydrogens, and likewise did not require a specified crystal structure to be used for pose or affinity prediction in Stage 1. Given this freedom to test a large number of crystal structures, especially for HSP90, many participants tested a number of workflows that went beyond a docking program’s sampling and scoring capabilities, and instead strove to incorporate target knowledge in an automated manner. This also allowed for expanding the challenge test beyond cognate ligand – receptor docking (known to be successful for the majority of docking algorithms) and challenge the community to test methods for the ability to handle water-mediated ligand-protein interactions, binding pocket flexibility, close chemotype (chemical series or similarity) docking and cross-docking. In the case of HSP90, care was taken to provide crystal structure examples of the binding mode for each of the three chemotypes, allowing for a similarity and cross-docking challenge. The MAP4K4 pose prediction challenge had an additional level of difficulty, in that there were no examples for binding modes for many of the compounds; on the other hand, MAP4K4 was, arguably, simpler in the sense that correctly treating water-mediated protein-ligand interactions was less critical than for HSP90.
2 Materials and Methods
2.1 Composition and Construction of Challenge Datasets
2.1.1 Raw Datasets
2.1.1.1 Heat Shock Protein 90
The HSP90 dataset used for this challenge is based on a large collection of enzyme inhibition data contributed by Abbvie Pharmaceuticals to CSAR, D3R’s predecessor which further developed the dataset by adding new compounds and binding data. The Abbvie dataset contains a set of small molecules with their IC50 values for binding to the protein’s N-terminal ATP-binding domain, measured with a time-resolved fluorescence energy transfer (TR-FRET) assay [14]. The dataset was expanded to a total of 180 with an additional set of 17 ligands, which were designed by the CSAR team, synthesized by WuXi AppTech and assayed by the same TR-FRET method. Some of the compounds were analyzed further by isothermal titration calorimetry and the OctetRed method [15], and had their pKas and solubilities measured. Although these additional data were not used herein, they are informative regarding the measurement uncertainties and are provided on our website (doi:10.15782/D6159W). Table S1 (SI-Dataset) provides the names, structures, and IC50 values for all compounds used in the present challenge. These compounds may be classified into three chemical series: aryl-benzimidazolones [16], pyrimidin-2-amines [14], and benzophenone-like compounds. Each series includes approximately 11 compounds for which binding was undetectable by the TR-FRET assay, corresponding to an IC50 greater than ~50μM (H.A. Carlson, personal communication). IC50 values ranged from 5.2 nM to >50μM. The CSAR team also obtained co-crystal structures using exactly the same truncated form of the protein used in the binding assays with eight of the ligands, as listed in Table S1 (SI-Dataset). Resolution limits for the resulting co-crystal structures range from 1.60 – 1.95 Å, and the crystallization methods and conditions can be found in www.RCSB.org. Some of the AbbVie compounds were further characterized by other assays as part of the CSAR effort, as detailed in the SI (HSP90_Materials&Methods; HSP90_Experimental_Data).
2.1.1.2 Mitogen-Activated Protein Kinase Kinase Kinase Kinase 4
The MAP4K4 dataset used here was contributed by Genentech, Inc., and comprises 30 crystal structures of the enzyme’s catalytic domain bound to 30 chemically varied ligands, together with IC50 values, measured by an ATP consumption assay [7] for 18 of the 30 compounds, and Ki values for 14 of the compounds determined by a Surface Plasmon Resonance (SPR)-based fragment screen[17], as summarized in Table S2 (SI-Dataset). The IC50 values ranged from 0.0031 to 10 μM. Additional assay results for a partial list of the compounds are available at the D3R website, doi:10.15782/D6WC7Z, providing some estimates of uncertainties.
2.1.2 Analysis and Refinement of HSP90 and MAP4K4 Datasets
Crystal structure coordinates were interrogated for quality of ligand and side chain occupancy around the binding pocket guided by both visual inspection of 2m|Fo|-D|Fc| difference Fourier syntheses generated using the MOE software package [18] and data quality estimates found in the wwPDB Validation Report (www.rcsb.org/validation/validation-reports; [19]) for each co-crystal structure. The most important criterion was the clarity of the difference electron density feature corresponding to each ligand, as shown in Tables S1 and S2 (SI-Dataset). The 2m|Fo|-D|Fc| maps contoured at 1.0σ clearly indicate the existence of the ligand in each co-crystal complex. For ligands of the HSP90 data set, the real space correlation coefficient (RSCC) ranges from 0.92 to 0.99 and the real space Rfactor (RSR) ranges from 0.05 to 0.1, leading to very low Zscores (RSRZ), ranging from −0.93 to 0.02, much lower than the cutoff value (ZRSR=2.0) for poor electron density; RSCC and RSR values generated by wwPDB DCC program [20]. For the ligands of the MAP4K4 data set, RSCC ranges from 0.89 to 0.98, RSR from 0.09 to 0.27, and RSRZ from 0.88 to 1.24 which are more diverse than the HSP90 cocrystal structures yet well below the cutoff for poor quality. Additional information is provided (see supplemental information: SI_Crystallographic_data_HSP90 and MAP4K4). The MAP4K4 crystal structure for ligand MAP32 was re-refined after the close of the challenge, to make the tautomeric state of the ligand pyrazole moiety more consistent with the observed hydrogen bonds the ligand makes between kinase protein backbone in the hinge region; this modification also required a 180-degree rotation of the pyrazole-phenyl bond. The RMSD values reported in this paper were evaluated against this new MAP32 model and differ somewhat from those previously distributed to challenge participants.
We also looked for possible consequences of crystal packing on ligand poses, as these might undermine the validity of these poses as references for pose predictions. The MAP4K4 crystal structures contain two protein copies per asymmetric unit, with only one copy containing bound ligand. Here, the bound ligands made interactions to only one polypeptide chain, so all 30 co-crystal structures were suitable for the challenge. However, the structure of HSP90 with ligand HSP90_44 (4YKT) was disqualified, because the bound ligand directly contacts a second molecule of HSP90, raising the likelihood of a significant perturbation of the pose, relative to what would exist in solution (SI-Figures, Figure S1). Additionally, two initially blinded HSP90 co-crystal structures were provided to participants as docking targets (see below), leaving five of the original eight pose-prediction challenges for this target.
2.2 Challenge Procedures
2.2.1 Posing the Challenge
For both the HSP90 and the MAP4K4 datasets, the challenge was held in two stages, as previously done by CSAR [4]. In Stage 1, participants could predict the poses of the ligands for which blinded crystal structures were available and also predict the rankings of all ligand binding affinities, or the binding free energies. Immediately after Stage 1 closed, all of the available co-crystal structures were released to the participants, and Stage 2 provided a second chance to predict or rank affinities, now with the presumed advantage of full access to all structural data.
Participants were provided with SMILES strings of the ligands to be docked, the pH at which the binding assays were performed, and the crystallization buffer conditions. Protein-ligand crystal structures were also provided, each of suitable quality for docking studies (overall resolution <2.5Å), and exemplifying relevant ligand binding modes, water-mediated interactions, and protein conformations, when available. The provided co-crystal structures were all translationally and rotationally aligned to a single reference structure within each dataset. These alignments were based on the coordinates of the α-carbon backbone atoms and emphasized secondary structure elements (α-helices and β-sheets) [18]. As mentioned above, the structures were provided to participants at the Challenge outset or as “Answers” after completion of Stage 1. Participant instructions permitted docking of ligands into other structures available in the PDB, and encouraged full utilization of information available in the scientific literature. The provided structures were chosen through in house docking and analysis of the active sites for both targets. For the HSP90 challenge, two crystal structures from the blinded dataset were provided at the outset of the challenge to exemplify new chemotype/water-mediated binding modes not found in the public domain. Where multiple crystal structures could have been selected, we chose to provide the structure refined using the highest resolution data. All challenge data were provided via the D3R website.
For pose predictions, participants were invited to submit up to five poses for each ligand, with one marked as their top-ranked pose. For affinities, participants were asked to submit a ranked list of all ligands for each protein target. In addition, three small (4, 5 and 10 ligands) sets of HSP90 ligands (Table 1) were identified as sufficiently similar to be amenable to explicit solvent alchemical free energy calculations [21], and were thus proposed as targets for this relatively computationally intensive approach.
Table 1.
The IDs and potencies of the HSP90 ligand sets in the free energy sets. Section 2.3.2 discusses the uncertainties in these data.
| ID | IC50 μM |
|---|---|
| SET 1 = 5 ligands | |
|
| |
| hsp90_80 | 1.91 |
| hsp90_81 | 0.206 |
| hsp90_82 | 11.6 |
| hsp90_83 | 15.0 |
| hsp90_84 | 5.85 |
| SET 2 = 4 ligands | |
|
| |
| hsp90_100 | 50.0 |
| hsp90_101 | 0.192 |
| hsp90_105 | 0.123 |
| hsp90_106 | 0.0874 |
| SET 3 = 10 ligands | |
|
| |
| hsp90_10 | 4.89 |
| hsp90_11 | 0.215 |
| hsp90_15 | 6.75 |
| hsp90_19 | 6.59 |
| hsp90_21 | 0.193 |
| hsp90_23 | 3.08 |
| hsp90_26 | 0.0205 |
| hsp90_28 | 0.276 |
| hsp90_34 | 6.14 |
| hsp90_61 | >50.0 |
For HSP90, Stage 1 opened Sep 15, 2015 and closed Nov 20, 2015; Stage 2 started several days after the close of Stage 1 and ended Feb 02, 2016. For MAP4K4, Stage 1 ran from Oct 16, 2015 to Dec 16, 2015; Stage 2 also started soon after and ran until Feb 02, 2016. Pose prediction answers for Stage 1 were released at the end of the Stage. As noted above, all available co-crystal structures were released immediately after the close of Stage 1 so they could be used in a second round of affinity predictions. No limit was placed on the number of predictions a participant could submit for each component of the challenge. Participants were provided the option of remaining anonymous and were able to change their choice in this regard until the experimental results were released.
2.2.2 Submission and Validation of Predictions
Participants were required to provide their predictions in defined formats, in order to enable automated processing of their submissions. Each ligand pose was submitted in the form of a legacy PDB format file, containing both ligand and protein coordinates, and a REMARK line with an energy or score for the pose. Although participants were free to dock the ligands into the protein structure(s) of their choice, the coordinates were required to be superimposed on those provided (above). Affinity rankings and free energy predictions were submitted in the form of comma-separated-value (CSV) files, with one ligand per line. Each submission was also required to include a protocol file, containing an informal name for the procedure used to predict the poses and/or affinities, the name(s) of the main software packages used, the values of key parameters, and a brief, plain-text description of the method. Detailed instructions, examples of correctly formatted submissions, and fillable template files, were provided to participants via the D3R website.
In order to participate in the challenge, a user or group was required to create a D3R login and then register for the challenge. Upon registration, the participants could access all available files to date, and upload predictions during open submission windows. Immediately after being uploaded by the participant, each new submission file was automatically scanned for a set of possible formatting and technical problems, and the submitter was immediately notified of any apparent problems. If no problems were detected at this stage, the submission was accepted and assigned a Submission ID. Each registered participant could then view his or her set of completed submissions, on a D3R page specific to the D3R login. Further validation checks of each prediction file took place subsequent to initial submission and assignment of the Submission ID. For example, a pose prediction was considered invalid if the PDB file contained the wrong ligand or if there were multiple copies of a ligand in one PDB file. Submitters were notified of any apparent problems at this stage and allowed to correct their submissions if they could do so before the close of the submission window.
Some submissions for the affinity ranking components of the challenges did not include predictions for all 180 ligands in the case of HSP90 or all 18 ligands in the case of MAP4K4. Those containing predictions for only two ligands were not analyzed. In the case of HSP90, some submissions contained rankings for only the six ligands in the pose-prediction part of this challenge; for these, the results were analyzed and provided to the submitters but were excluded from the analysis in this report.
2.3 Evaluation of Predictions
2.3.1 Evaluation of Ligand Poses
The accuracy of each predicted ligand pose was evaluated in terms of the root-mean-square deviation (RMSD) of the prediction relative to the crystallographic pose. Because submitters were instructed to provide coordinates aligned with the structures provided as references, the structures were not adjusted in order to compute the RMSD values. However, we used the maximum common substructure functionality of the OEChem Python toolkit [22] to correct for possible renumbering of ligand atoms and for ligand symmetries, as previously described [23]. Although participants were free to treat the protein as flexible, we did not evaluate the accuracy of the protein conformation, or of water molecules, in the pose predictions. As noted above, participants were invited to submit up to five poses per ligand, with one of the five identified as top ranked. We computed the RMSD values of all poses in each submission, and reported statistics on both the top ranked pose, and, separately, the best (lowest RMSD) pose among the five. The results for HSP90 and MAP4K4 were analyzed and are presented separately, rather than merging results by methodology, partly because it was impossible to be sure which methods (if any) were identical from one target to the other; and partly because, as detailed below, target-specific issues other than the docking and scoring method itself proved to be important determinants of docking accuracy.
2.3.2 Evaluation of Affinity Rankings
Predicted affinity rankings were evaluated in terms of the Kendall’s tau and Spearman’s rho rank correlation coefficients. Both range from 1 to −1, where 1 indicates a perfect ranking and −1 represents a perfectly reversed ranking. The Results section reports Kendall’s tau and Spearman’s rho for the predictions as submitted (i.e., without resampling), along with uncertainties from the following resampling procedure. The uncertainty in each correlation coefficient was assessed over 10,000 rounds of bootstrap resampling with replacement, where, in each round, the experimental IC50 data were randomly modified based on the experimental uncertainties and reranked accordingly. The modified IC50 values were generated by converting each IC50 into a free energy, ΔG = −RTln (IC50), (T=300K), adding a random offset δG drawn from a Gaussian distribution of mean zero and standard deviation RTln(Ierr), and then converting back to an IC50 as IC50′ = e−(ΔG+δG)/RT. The value of Ierr was set to 3 for HSP90 based on published estimation of assay accuracy at Abbvie [24] and comparison of assay data for 17 compounds where the TR-FRET assay and ITC Kd values had a median difference of 2.09-fold and average of 3.47 (see D3R website). The value of Ierr for MAP4K4 was set at 2 based on evaluation of assay results for 15 compounds measured in the same assay at two different locations and the two different assays all have median differences less that 2-fold (see D3R website).
Some submission files included multiple variants of the same method. In order to simplify reporting, we report only the highest score from each such submission; the names of the methods for such submissions have number of prediction sets in the method name (parenthesis), in Tables S4–S7. The numbers of rankings before and after these mergers are reported in Table 1.
Eight compounds (HSP90_35, 61, 94, 95, 116, 123, 127 and 170) have chiral centers and the affinities were reported as pertaining to racemic mixtures. Recalculation of all Kendall’s tau statistics without these compounds led to minimum, mean and maximum changes in this statistic of −0.02, 0.01 and 0.04. This did not change the trends and the reported results include these compounds.
Two simple null models [25] were set up as trivial performance baselines and evaluated in the same manner as the submitted predictions. The null models are “Mwt”, in which the affinities were ranked by molecular weight; and clogP [26] in which affinities were ranked based on the octanol-water partition coefficient computationally calculated by CDD Vault [27].
2.3.3 Evaluation of Free Energy Predictions
Participants were invited to submit predictions of relative binding free energies for three small sets of chemically similar HSP90 ligands. Only IC50 measurements are available for these compounds. Although IC50s can approach dissociation constants, our application of the Cheng-Prusoff equation [28] for the HSP90 TR-FRET assay conditions suggest the relationship of Kd to IC50 is ~ 1:3 to 1:5 ([Geldanamycin] = 0.8μM, 3 hour incubation and Kd ~ 20 – 50nM). However, a constant ratio of this sort will not perturb relative binding free energies, so the relative energies can be compared with experiment. Because each set includes compounds with similar affinities (Table 1), even small errors in the computed free energies can lead to reranking of the ligands. Ranking metrics like Kendall’s tau may therefore be overly sensitive to quantitatively small errors and thus be uninformative. Therefore, although Kendall’s tau is reported for these special sets, we focus more on the quantitative accuracy of the results, using the centered root mean square error (RMSEc) of the calculated binding free energy differences versus those from experiment [29, 30]:
| (1) |
Here N is the number of ligands in each set (4, 5 and 10), and ΔΔGcalc and ΔΔGexpt are the submitted and experimental binding free energies of ligand i relative to one arbitrarily selected ligand, here termed ligand 0. The advantage of the RMSEc over other statistics that might be used to characterize the accuracy of computed relative binding free energies is that it does not depend on which ligand is selected as the reference ligand.
The free energy component of the HSP90 challenge centered on three small sets of chemically similar compounds. However, some free energy submissions included all 180 compounds for HSP90 or all 18 compounds for MAP4K4. Only one of these submissions used the explicit solvent free energy methods that were envisioned for the free energy challenge, and this submission was subsequently withdrawn by the participant. The remaining all-ligand free energy submissions were merged into and analyzed with the ranking submissions. Note, however, that a number of methods other than explicit solvent free energy simulations were applied to the small free energy sets, and these are included in the comparison of free energy methods.
3 Results
A total of 38 participants uploaded a total of 355 submissions that passed validation tests (above). Note that 11 out of these 355 submissions did not include the method names and protocols used and are not included in this paper. The numbers of valid submissions for the seven components of the challenge are listed in Table 2. The methods used ranged from knowledge-based to physics-based, and include both automated and manual methods. The methods are summarized in Tables S3 – S9 (SI-Methods), and further details may be found in papers from the participants, most or all of which are published in the same special issue as the present overview article. The following subsections analyze the performance of these methods for ligand pose prediction and the assessment of ligand binding potency.
Table 2.
Number of validated submissions, Nsubmit, received for each component of D3R Grand Challenge 2015. The numbers in parentheses for the affinity rankings are the numbers of “merged” submissions, as explained in Section 2.3.2.
| Challenge Component | Nsubmit |
|---|---|
| HSP90 Pose Predictions | 47 |
| MAP4K4 Pose Predictions | 33 |
| HSP90 Stage 1 Affinity Ranking | 75 (41) |
| MAP4K4 Stage 1 Affinity Ranking | 77 (40) |
| HSP90 Stage 2 Affinity Ranking | 59 (30) |
| MAP4K4 Stage 2 Affinity Ranking | 46 (26) |
| HSP90 Small Set Free Energies | 18 |
3.1 Pose Predictions
3.1.1 HSP90 Pose Predictions
3.1.1.1 Overview of Prediction Accuracy
The results for 39 sets of HSP90 pose predictions submitted for all five ligands along with method details are summarized in Figure 1, in terms of RMSD statistics for each submission (Table S3, SI-Methods) over the five ligand structures, with variances in the RMSDs across ligands are illustrated in terms of boxes and whiskers. This presentation, though perhaps not needed for such small datasets, facilitates comparison with analogous graphs for the larger MAP4K4 study. The methods are ordered along the horizontal axis by the median RMSD of the submissions’ top ranked poses (rank 1). The left-hand panel provides statistics for these rank 1 poses, while the right-hand panel shows results for the lowest RMSD poses across up to five poses submitted for each ligand (best of top 5). The median RMSDs of the rank 1 poses range from 0.3 to 6.6 Å, and the corresponding range for the best of top 5 poses is essentially the same. However, the rank 1 poses were the best of the five 56% of the time, which is better than the expected fraction of 20% if the ranking were entirely random.
Figure 1.
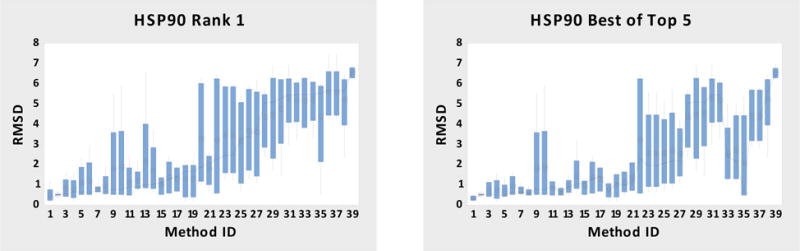
HSP90 RMSD box plots of rank 1 and best of top 5 poses for all submitted-methods. The methods are ordered by the median RMSD. The mean is shown by the circles, the connect line is for the median, the box is for the interquartile range (IQR) with the whiskers indicating 1.5X IQR. Table S3 (SI-Methods) has the names of the Submitted Methods and a summary of the protocols. Each Submitted Method’s box plot contains 5 data points.
3.1.1.2 Correlation of Performance with Docking Software and Method
Half of the submissions provided rank 1 poses with median RMSD < 2Å, and thus met a reasonable and common criterion of success. However, it is not immediately obvious that the success of these submissions can be attributed to the choice of docking software, as they used a range of tools (rDOCK, AutoDock Vina or a variant thereof, Gold, and Surflex) as well as some combinations (Gold-PlantsPLP-rDock, RosettaLigand-Omega-ROCS, Surflex-Grim and Glide-Prime-Desmond-Qsite). Additionally, submissions using similar or the same software packages yielded differing levels of accuracy; for example, methods using AutoDock Vina and Glide are scattered through the ranking. It is suggestive that four of the most successful 11 methods mention visual inspection of computationally generated poses, while apparently none of the 28 less successful methods included human intervention.
The RosettaLigand submissions provide an informative illustration of the complexities that arise in interpreting the present results. For method 4, the median RMSD of the rank 1 poses is only 0.5 Å; for method 25, this statistic is considerably worse, at 3.3 Å. Neither method used docking as conventionally interpreted; instead, both generated multiple ligand conformations and used superposition software to find an optimal overlay on the pose of a similar ligand with an available co-crystal structure, and RosettaLigand scores were determined for the resulting poses. In method 4, ligand conformations were generated with the program Omega [31], and the programs PoPSS [32, 33] and ROCS [34] were used for the overlay. In method 25, MOE and the in-house unpublished BioChemicalLibrary (BCL) tool were used.
3.1.1.3 Correlation of Pose Prediction with Protein Conformation and Binding Site Water Molecules
Further examination of the results suggests that docking success is influenced by the choice of protein structure and the treatment of binding site water molecules, perhaps more so than by the choice of docking software. All the HSP90 ligands fall into three chemical classes: benzimidazolone (ligand HSP90_40); aminopyrimidine (ligands HSP90_73 and HSP90_179); and benzophenone-like (ligands HSP90_164 and HSP90_175), and the protein crystal structures provided for this challenge included at least one determined with a ligand from each chemical series: 4YKR for benzimidazolone, 2JJC and 2XDX for aminopyrimidine, and 4YKY for benzophenone-like. However, the correspondence of protein structure to ligand class was not revealed, and participants were also free to dock these ligands into other HSP90 structures drawn from the PDB. We conjectured that predictions in which a ligand was docked into a structure solved with another ligand of the same congeneric series (“similarity docking”) might be more accurate that predictions in which a ligand was docked into a structure determined with an entirely different ligand (“cross-docking”). This conjecture is borne out for all three ligand classes, most clearly for the benzimidazolone and benzophenone-like classes (Figure 2). Note that each bar corresponds to 11–28 different docking submissions, providing a reasonable sampling of each approach.
Figure 2.
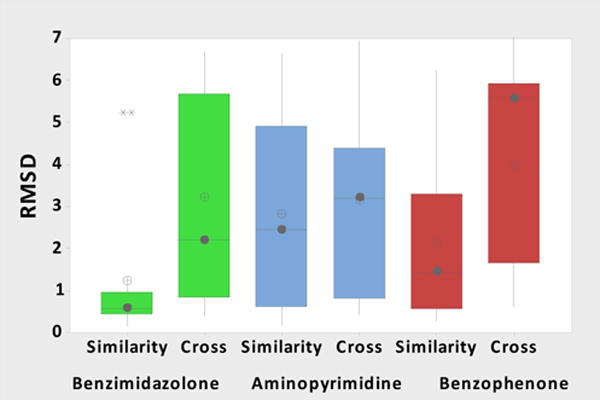
HSP90 box plot distributions for the rank 1 pose, color coded by chemical series and organized by receptor-ligand PDB selection type, “similarity” or “cross-docking”. The mean is shown by the circles with a plus sign, the median by the filled circle, the box is for the interquartile range (IQR) with the whiskers indicating 1.5X IQR. The outliers are indicated by asterisks above the whiskers. The box plots contain 12, 27, 24, 15, 11, and 28 data points, respectively.
In the above plot, “similarity” refers to the listed chemical series chemotypes, rather than to overall Tanimoto similarity. Chemotype similarity can assist in selection of crystal structures with similar chemical series that provides a side chain (and water) template for binding mode within a chemical series but not necessarily the best binding pocket conformation, particularly where there is binding site flexibility. Participants that defined similarity more generally across the entire ligand could select structures that accommodate the need for a more open ATP lid structure, Figure 3. Examination of the two aminopyrimidine ligand co-crystal structures (Figure 3) reveals ligand HSP90_73 binds to an open conformation (yellow), while ligand HSP90_179 binds to a closed conformation (purple), and that the nitro group of HSP90_73 would have a steric clash with the closed form.
Figure 3.
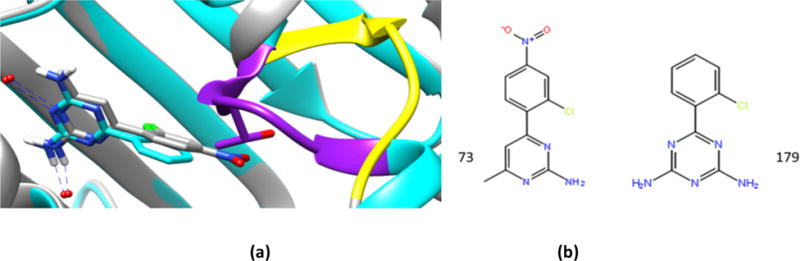
Binding site conformations of of HSP90 (a), with ligands 73 (grey) and 179 (cyan) in the experimental co-crystal structures. The open conformation with ligand 73 is shown in yellow while 179’s closed conformation is shown in purple, with the positioning of Thr109 depicted. (b) Chemical structures of the two ligands: 4YKW (HSP90_73) and 4YKU (HSP90_179), respectively.
For the benzophenone-like chemotype, similarity could assist with binding mode prediction (Figure 4), but participants had more difficulty predicting the pose of HSP90_175 (median RMSD 5.7 Å) versus ligand HSP90_164 (median RMSD 1.8 Å). We find that the protein structures used by the participants influence accuracy in different ways for these compounds, despite having similar binding modes (Figure 5). Both experimental co-crystal structures have the same closed conformation of the protein, and the most notable difference is a water-mediated interaction for ligand HSP90_175 (absent for HSP90_164), although the water position is identical in both cases (Figure 4). One might therefore expect omitting the water molecule during docking for ligand HSP90_175 to be a problem, but not necessarily for HSP90_164. (Note that one of the protein structures provided for the docking exercise, 4YKY, was determined with a ligand of this chemotype and has the appropriate closed conformation and the conserved waters present.) Indeed, as shown in Figure 5, predictions for ligand HSP90_175 that used a closed conformation and included the water molecule had the best results, while docking without the water molecule led to high median RMSDs, regardless of loop conformation. (None of the participants used an open conformation structure with the water molecule present.) However, the presence or absence of the water molecule mattered less for ligand HSP90_164 (Figure 5): with or without the water molecule, using the correct conformation (closed) of the receptor resulted in low median RMSDs, and a high median RMSD was observed only when incorrect conformation (open) was used in the absence of the bound water, perhaps by restricting the sampling space for the possible binding modes. It is remarkable that many of the most successful workflows used superposition of “similar” ligands and avoided the sampling of large binding site spaces.
Figure 4.
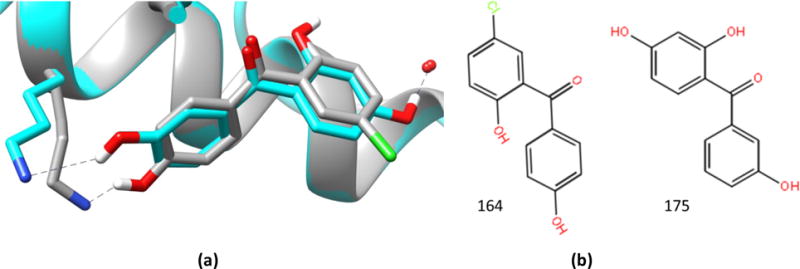
(a) Ligands 164 (grey) and 175 (cyan) in the experimental co-crystal structures. (b) Their 2D structures; 4YKX (HSP90_164) and 4YKZ (HSP90_175), respectively.
Figure 5.
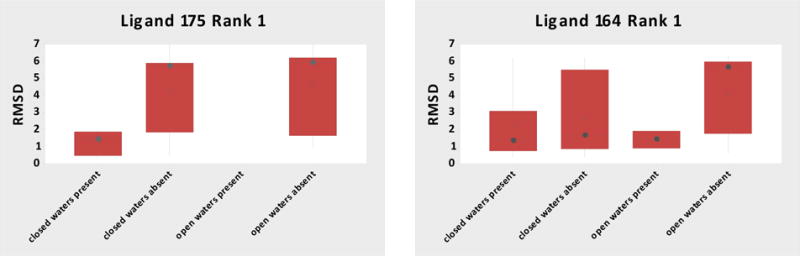
HSP90 box plot of RMSD distributions for rank 1 poses of ligands HSP90_175 and HSP90_164, separated according to the conformation of the protein structure used and whether the crucial water was present or absent in the docked structure. (An open conformation and the water-present structure was not tested for ligand HSP90_175.) The means are shown by circles with a plus sign, the medians by the filled circles, the boxes are for the interquartile ranges (IQR), and the whiskers indicat 1.5 × IQR. The respective box plots contain 6, 21 and 13 predictions for ligand 175; and 8, 17, 2, 12 predictions for ligand 164.
3.1.2 MAP4K4 Pose Predictions
3.2.2.1 Overview of Prediction Accuracy
The results for the 30 sets of MAP4K4 pose predictions are summarized in Figure 6, which shows RMSD statistics for each submission (Table S3) over the 30 ligand structures, with variances across ligands expressed in terms of boxes and whiskers. Overall, these predictions are considerably less accurate than those for HSP90 (Figure 1). Only one out of 30 submissions has a median RMSD below 2.0 Å for rank 1 poses, compared to 20 out of 39 for HSP90. Furthermore, for MAP4K4, the median RMSDs for rank 1 poses range from 1.6 to 8.8 Å, while the range for HSP90 is 0.3–6.6 Å. Nonetheless, it is encouraging that the rank 1 pose is the best of the five submitted poses for 52% of the submissions, much more often than the 20% which would be expected if the rankings were random (as seen in the HSP90 challenge).
Figure 6.
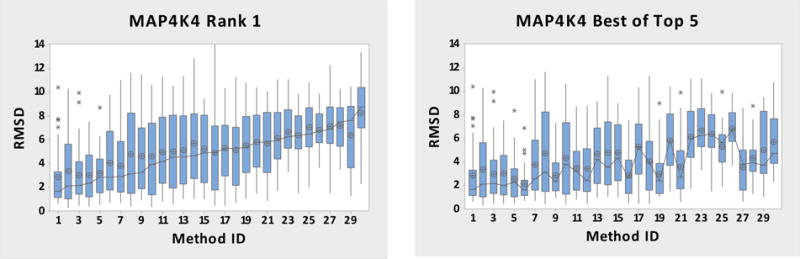
MAP4K4 RMSD box plots of rank 1 and best of top 5 poses for all submitted-methods. Means are shown by circles, the connecting line is for the medians, the box is for the interquartile range (IQR), and the whiskers indicate 1.5 × IQR. Outliers are indicated by asterisks above the bars. Table S4 (SI-Methods) has the names of the Submitted Methods and a summary of the protocols. Each Submitted Method’s box plot contains 30 data points.
The MAP4K4 pose prediction challenge was anticipated to be more challenging than HSP90 for a number of reasons. With respect to cross-docking, there were far fewer relevant co-crystal structures available in the PDB (eight versus > 200 for human HSP90). Available MAP4K4 co-crystal structures exemplified limited diversity in bound ligand chemotypes and the range of chemotypes in the dataset was highly diverse. Another factor noted by a number of participants was the potential for a large binding site size, depending on the conformation of the glycine-rich P-loop [13].
3.1.2.2 Correlation of Performance with Docking Software and Method
For MAP4K4, the only submission with a median RMSD less than 2.0 Å for the rank 1 poses used Method 1, named Glide SP-Qsite. Two additional submissions achieved a median RMSD less than 2.0 Å for the best of top 5 poses; these used Method 4 and 6, Vina and RosettaLigand-Omega-PoPPs-ROCS respectively. As noted above, method 6 is not a true docking method, but instead is based on superposition of the ligand to be docked on the pose of a ligand with an available co-crystal structure. These three approaches were among the more accurate ones used for HSP90. However, much as observed for HSP90, the other RosettaLigand methods, which here used docking rather than another overlay method, were not as predictive. In fact, just as for HSP90, submissions based on a given piece of software could provide widely ranging performances, depending on the details of how the software was used. Thus, methods based on Glide, RosettaLigand and AutoDock Vina appears throughout the rank list of methods (Table S4). For a discussion of methods that appear to provide relatively accurate performance across both datasets, see Section 3.1.2.3, below.
3.1.2.3 Role of the Protein Conformation Used for Pose Prediction
For HSP90, “similarity docking”, in which each ligand was docked into a protein structure solved with another ligand of same or similar chemotype, tended to be more predictive than true cross-docking into a less-related protein structure, as noted above. This strategy was less successful across the diverse range of chemotypes presented in the MAP4K4 dataset. Four submissions (4, 10, 13 and 22) that used ligand similarity-based structures for selection of the protein target had median RMSDs of 2.7, 4.6, 5.0 and 6.5 Å respectively, when all compounds were considered. However, ten compounds (one third of the dataset; compounds 3, 14–16, 18, 19, 21, 22, 25, & 27) have the closely related aminopyrimidine/aminoquinazoline chemotypes of published structures [7]. When the evaluation is limited to these compounds the median RMSD values for these four submissions are 1.6, 2.7, 2.6 and 6.1 Å, respectively. Inspections of pose prediction on a per compound basis (SI figures, Figure S2) illustrates that nine of the 15 compounds with the lowest medians are in this aminopyrimidine/aminoquinazoline class. Notably, one of the better performing methodologies expanded the number of crystal structures by considering chemotype similarity to closely related kinases with more diverse sets of bound ligands (Table S3, protocol 4).
Compounds that had no similarity to common chemotypes known to bind the kinase hinge region presented a significant pose prediction challenge, namely the 3 benzoxepins (MAP12, MAP13 and MAP17 gold colored in Figure S2). These cases were challenging for two reasons. They have a difficult-to-predict 7-membered ring conformation, and they lack obvious kinase hinge binding hydrogen bond donors and acceptors. Moreover, MAP17 presents a particularly difficult case as it does not have any direct hydrogen bonding to the pocket and its interaction with the kinase hinge backbone is water-mediated.
Interestingly, the level of difficulty in these pose predictions does not appear to correlate with potentially relevant ligand properties, such as molecular weight, presence of tautomers, or number of rotatable bonds; with features of the protein structures used for docking, such as the conformation of the P-loop (open versus closed) or the crystal structure resolution; or with the ligand-protein binding affinity.
3.1.3 Performance of Methods across Both HSP90 and MAP4K4
Certain well defined methods were applied to both the HSP90 and MAP4K4 pose prediction challenges, allowing us to assess their predictive ability across targets. We focused on the more predictive 20% of submissions for each target, which corresponds to the top ranked eight for HSP90 and five for MAP4K4. The methods in common between both lists are DockBench, which appears second on both lists, and which, as part of its procedure, analyzes which of a number of docking methods best works for given targets and in this case selected Gold, PlantsPLP and rDock; Surflex-GRIM, which appears third in both lists; and RosettaLigand-Omega-ROCS, which appears fourth in both lists, and, as noted above, is a ligand-overlay method rather than a full-docking method. As noted above, the best performing methods for pose prediction were less associated with a single docking algorithm than with a “similarity-docking” approach.
3.2 Predictions of Ligand Binding Potency
This section examines the reliability of potency predictions in terms of the ability to correctly rank-order the full sets of HSP90 and MAP4K4 ligands, and free-energy-based methods to rank order a subset of HSP90 ligands.
3.2.1 Affinity Rankings
3.2.1.1 Overall Evaluations
Participants were asked to predict the ranking of affinities for 180 HSP 90 ligands and 18 MAP4K4 ligands. This challenge component was run in two stages. During Stage 1, none of the co-crystal structures of these ligands with their respective targets were available to the participants. In Stage 2, participants had a second opportunity to rank-order all of the ligands by affinity, this time with knowledge of all of the co-crystal structures available (i.e., five structures for the HSP90 ligands and all 18 for the MAP4K4 ligands). The results, summarized in Figure 7 and Figure 8, focus on the Kendall’s tau statistic, with error bars indicating one standard deviation in the bootstrapping analysis (see Methods). The Spearman’s rho results added little information (Figures S3 and S4).
Figure 7.
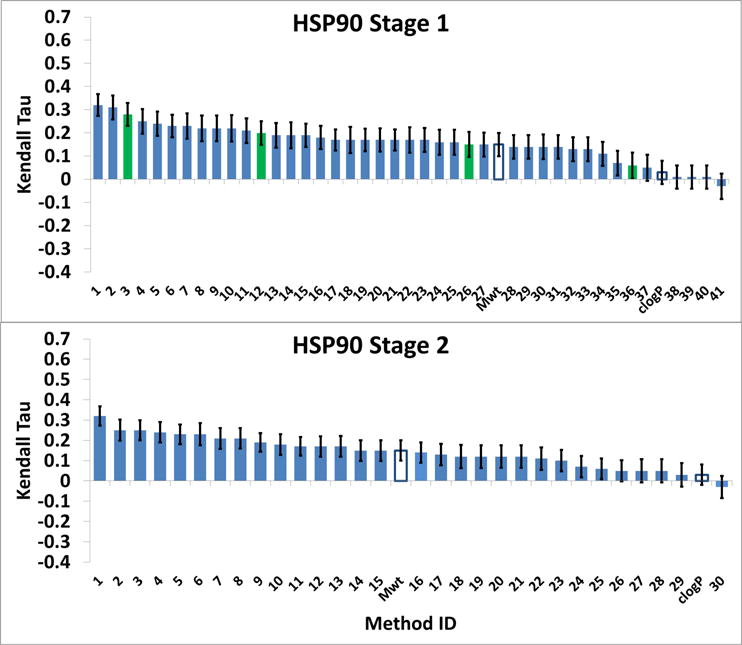
HSP90 Kendall Tau correlation coefficient scores between the predicted scores and experimental binding affinities. Green bars are for ligand-based scoring methods, and unfilled bars are for null models. The method names corresponding to the Method IDs are in Tables S5 and S6 (SI-Methods). The error bars are 1σ confidence intervals based on 10,000 bootstrap samples.
Figure 8.
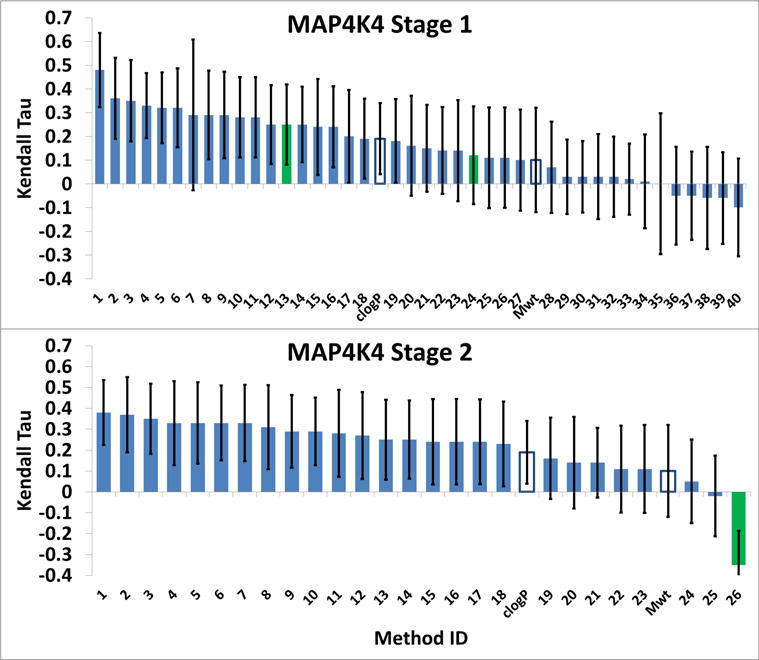
MAP4K4 Kendall Tau correlation coefficient scores between the predicted scores and experimental binding affinities. The green bars are for ligand-based scoring methods, and unfilled bars are for null models. The names corresponding to the Submitted Method’s number are in Tables S7 and S8 (SI - Methods). The error bars are 1σ confidence intervals based on 10,000 bootstrap samples. They are fairly large for the MAP4K4 dataset due to a relatively big experimental uncertainty.
Almost all of the submitted rankings correlate positively with the experimental ranking (Figure 7 and Figure 8), with mean and median tau values of 0.15 and 0.17, respectively, for HSP90, and 0.18 and 0.24, respectively, for MAP4K4. These results are statistically meaningful, given that the standard deviation of the tau values provided by resampling (see Methods) are all in the range 0.046–0.057 (mean 0.052), and indicate that a range of current methods have predictive value for ranking ligand affinities. On the other hand, the correlations are not particularly high, with maximum values of about 0.32 for HSP90 and 0.48 for MAP4K4. For comparison, an ideal computational method that yields results in exact agreement with the experimental IC50s, would have Kendall’s tau values of 0.76±0.02 and 0.80±0.07, for HSP90 and MAP4K4, respectively, after boostrap resampling accounting for the experimental uncertainties. Another baseline reference for the predictions is provided by the null models; ranking by molecular weight and clog P. These yield positive correlations with experiment, but neither null model is particularly accurate, as their tau values fall near or below the median of the predictions. In addition, whereas molecular weight did better than clog P for HSP90, clog P did better for MAP4K4, and neither did well for both systems (Figure 7 and Figure 8).
Perhaps surprisingly, information about ligand poses did not lead to more accurate affinity rankings. Thus, the rankings are about equally accurate for HSP90 and MAP4K4 in Stage 1, even though pose predictions for the former tended to be more accurate. Moreover, access to the known crystallographic poses in Stage 2 did not improve the ranking results over Stage 1, even for MAP4K4, where co-crystal structures were provided for all 18 ligands to be ranked. Also notable is that purely ligand-based methods (green bars in Figure 7 and Figure 8), which do not use information about the protein structure, were not clearly distinguishable from the structure-based methods, as they exhibited a wide range of performance, from near the best in HSP90 Stage 1 to the worst in MAP4K4 Stage 2.
In order to identify submissions that gave above average performances, we consider the uncertainties in the values of tau. For HSP90 Stage 1, the mean value of tau is 0.159, and the standard deviations of all the tau values are in the range 0.046 – 0.057, so we use the mean, 0.052 for all. The standard deviation of the difference between two tau values then obtained by adding the two standard deviations in quadrature, and one may add the result to the mean of tau to find that a tau of 0.306 is a convincing two standard deviations above the mean. Method 1 (vina-smina(7), tau 0.32) and Method 2 (rdock(2), tau 0.31) meet this criterion, and Method 3 (qsar(4), tau 0.28) is close. Applying the same criterion to HSP90 Stage 2 yields only Method 1 (vina-smina(7), with tau of 0.32. For both stages of MAP4K4, the uncertainties in tau are much larger (means of 0.19) because the number of data points is 10-fold smaller. Applying the same criterion as for HSP90 yields only one prediction at least two standard deviations above the median, Method 1 (PLANTS + Pyplif_subset-Vina) in Stage 1. Unfortunately, this method does not appear to have been applied to Stage 2 or to HSP90, so no consistency check is available.
The vina-smina (7) submission that did well in the HSP90 rankings included seven variant sub-methods, and we have focused here on only the top performing variant. Encouragingly, it is the same variant that did best for both stages of HSP90, and this also yielded tau values of 0.29 and 0.29 for Stages 1 and 2 of MAP4K4, which are above the means. The method involved generating ligand conformers with the program Omega [31] aligning conformers to the most similar co-crystal ligands in the PDB, minimizing the aligned conformers in the co-crystal binding site, and recording the highest docking score obtained.
3.2.1.2 HSP90 Affinity Rankings by Chemotype
We considered whether it was easier to rank the affinities of a series of compounds with a common chemical scaffold, as opposed to a heterogeneous set. The 180 HSP90 ligands were classified into three chemotype (CT) groupings by CSAR: benzimidazolone (CT1, 61 compounds, IC50 0.0052 to 42 μM), aminopyrimidine (CT2, 62 compounds, IC50 0.016 to 50 μM), and benzophenone (CT3, 57 compounds, IC50 0.01 – 50 μM). The rankings of the benzimidazolones (CT1) (Figure 9, top) are clearly better than the rankings of the full set of HSP90 ligands (Figure 7), as the highest tau values are about 0.51, rather than 0.32. On the other hand, the aminopyrimidine rankings (Figure 9, middle) are only marginally better (maximum tau about 0.38) than those for the full set, while those for the benzophenones (Figure 9, bottom) are somewhat worse (maximum tau about 0.22). These subset results, when compared with the “ideal” mean tau values of 0.77, 0.74 and 0.71, based on bootstrap resampling for the respective compound series, suggest that it was significantly easier to rank compounds in the benzimidazolone series, and harder to rank the benzophenones. The fact that the benzophenones are particularly problematic may have to do with their having lower molecular weights or generally weak affinities, relative to the other series. Also we note that this chemical series has more chemotype diversity than the others (see SI-Datasets).
Figure 9.
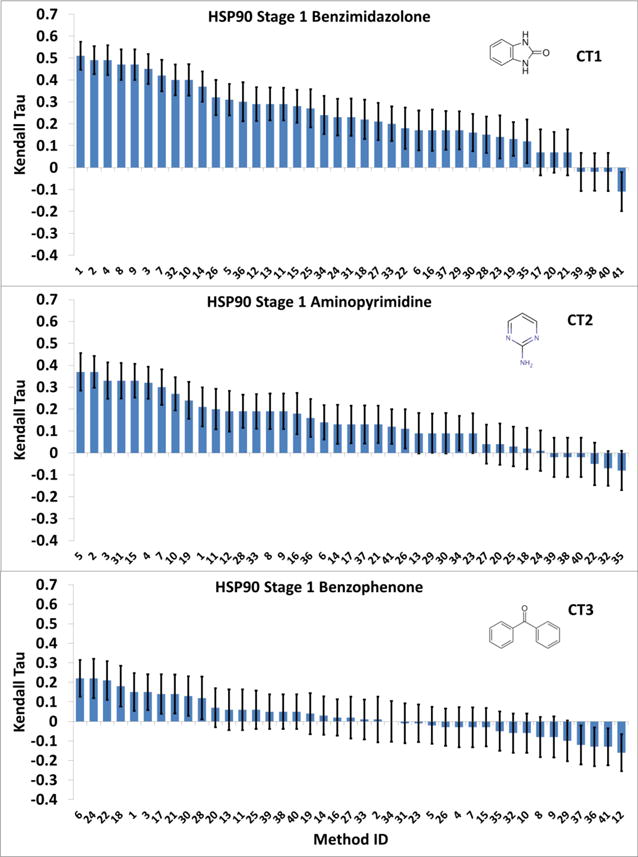
HSP90 Kendall Tau correlation coefficient scores between the predicted scores and experimental binding affinities, separated by the three chemotypes. The names corresponding to the Submitted Methods number are in Table S5. The error bars are 1σ confidence intervals based on 10,000 bootstrap samples.
3.2.2 Free Energy Predictions
In order to facilitate participation of research groups wishing to use this Grand Challenge to evaluate computationally intensive “alchemical” [21] methods of predicting differences in binding free energy, we included a challenge component in which three sets of chemically similar HSP90 ligands were put forward as targets for relative binding free energy predictions. Free energy Sets 1, 2 and 3 respectively comprise 5, 4 and 10 ligands and the binding free energies within each set span 2.6, 3.8 and 2.1 kcal/mol. Eleven predictions (Table S8) were submitted for free energy Sets 1 and 2, and ten were submitted for Set 3. Of these, three predictions in fact used alchemical free energy methods with explicit solvent, while the rest use methods based on analysis of small numbers of local energy minima generated by docking, with free energy estimates based on scoring functions, force fields with implicit solvent, and electronic structure calculations with implicit solvent. Two of the three predictions, Methods 5 and 11, used the same computational protocol; however, Method 5 was submitted at Stage 1 and omitted Set 3, while Method 11 was submitted at Stage 2 and include not only Sets 1 and 2 but also 9 of the 10 compounds in Set 3. Figure 10 and Table S8 summarize the results and methods in terms of RMSEc and Kendall’s tau; the Method IDs are ordered in terms of the average RMSEc across all three Sets, so that Method 1 has the lowest mean error and Method 11 the highest.
Figure 10.
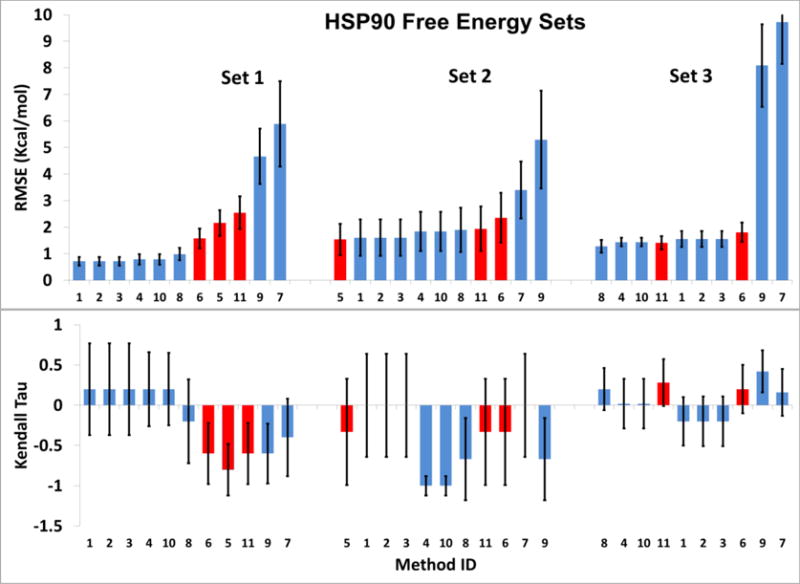
RMSEc (top row) and Kendall’s tau (bottom row) for the three free energy prediction sets. Methods using explicit solvent alchemical free energy simulations (5, 6 and 11) are shown in red. The X-axis labels are the Method IDs from Table S9, and are in order of increasing average RMSEc across all three sets. Error bars indicate 1σ ranges based on 10,000 bootstrap samples.
A number of methods provide errors (RMSEc) across the sets of roughly 1–2 kcal/mol (Figure 10, top row; Table 9). Most of the more accurate results come from methods based on docking (Figure 10, blue bars). Thus, Methods 1–3, which seem closely related to each other, as well as Methods 4 and 10, used scoring functions related to AutoDock and AutoDock Vina, while Method 8 is force-field based. The alchemical free energy methods (Figure 10, red bars) perform no better than the simpler docking-based methods, and perhaps somewhat worse in the case of Set 1. It is worth noting that the three sets of alchemical predictions are of similar accuracy: the bootstrap analysis suggests an expected accuracy range of about 1.0 – 3.3 kcal/mol. Finally, Method 7, which used electronic structure calculations instead of a scoring function or force field, yields particularly high errors of 3.4 kcal/mol (Set 2) to 9.7 kcal/mol (Set 3).
The Kendall tau statistics (Figure 10, lower row; Table 9) are associated with large error bars, presumably due in large part to the fact that each ligand set includes only a few compounds, and some of these compounds have quite similar affinities, which makes ranking difficult. Nonetheless, some of the predictions appear to yield fairly robustly reversed rankings, notably the alchemical methods and Method 9 for Set 1, and Methods 4 and 10 for Set 2.
4 Discussion
The D3R Grand Challenge 2015 attracted international participation and allowed a range of computational methods to be evaluated for prospective prediction of ligand-protein poses, ranking of ligands by affinity for a targeted protein, and prediction of relative binding free energies. The challenge was entirely open, in the sense that participants could use any and all existing information on the HSP90 and MAP4K4 systems, such as existing co-crystal structures in the PDB and affinity data available in articles and databases. In particular, for the pose-prediction component, participants could dock the ligands into HSP90 and MAP4K4 structures of their own choosing, rather than using structures provided specifically for this purpose, and affinity rankings could use structure-based and/or ligand-based methods.
Pose predictions for HSP90 were somewhat successful, as multiple submissions had median RMSD values well below 2.0 Å. MAP4K4 proved to be much more challenging, as few methods met this criterion. The higher success rates for HSP90 may indicate that it is an intrinsically easier system to model, but may also stem from the far greater number of HSP90 co-crystal structures in the PDB, which participants could use in various ways to enhance their methods. In fact, for HSP90, we observed a trend – though not a rule – that pose predictions were more accurate if they docked each ligand into a structure that had been determined with a ligand of the same chemical class. For some ligands it was also important to explicitly include certain crystallographic water molecules. A similar trend is observed for MAP4K4 for the ten compounds belonging to the chemical class(es) where co-crystal structures were publicly available; thus, participants did notably better at predicting poses for the aminopyrimidine/6-quinazolone class than the other diverse chemotypes in the dataset.
A closely related finding is that a given docking program could yield either high or low accuracy, depending upon extrinsic factors such as which protein structure was used for the docking, how protein structures were prepared, and other aspects of the protocol. This finding indicates that successful prediction of ligand-protein poses relies not only on the docking program, but also on other steps in the overall protocol, or workflow, which may include a docking program as a central component but also contains key preparatory or procedural steps. For example, some of the better performing methods in this challenge started by identifying an available co-crystal structure solved with a ligand similar to the one whose pose was to be predicted. Given such a well-chosen structure, a number of docking programs could then provide an accurate pose, if not as the top rank, certainly in the top five poses. One may draw an analogy to the case of comparative protein structure modeling, where accuracy hinges not only on the modeling technology used, but also on the availability of protein structures with similar sequences and on the quality of the method used for sequence alignment [35]. It also appears that multiple docking programs effectively sample relevant poses, as the better performing methods used various docking algorithms, a result supported by the success of cognate docking [2]. This analysis also supports the utility of integrated docking workflows, such as OpenEye’s POSIT [36] which systematically include knowledge-based steps to improve cross-docking.
The challenge of ranking ligands by affinity proved more difficult, consistent with other recent studies [37]. Although it is encouraging that nearly all predicted rankings yielded positive correlations with experiment, even the best predictions remain far from ideal. Of greater concern was the fact that prediction accuracy did not improve on going from Stage 1 to Stage 2. Thus, much of the ranking error must be attributed to the scoring or energy functions in use, not pose prediction. This holds particularly in the case of MAP4K4, where participants in Stage 2 knew the crystallographic poses of all 18 ligands whose affinities were to be ranked. One way to seek improved scoring accuracy is to use more detailed calculations, such as alchemical free energy simulations [21] which have recently provided encouraging results in protein-ligand modeling [38, 39]. The free energy component of Grand Challenge 2015 accordingly included three small sets of chemically similar ligands amenable to such calculations. However, these more rigorous methods did not yield improved accuracy, when compared with the results of several simple scoring functions that also were applied to these small free energy sets.
We also note that some of the challenge in ranking may be associated with the dataset itself. For example, several HSP90 inhibitors have time dependent binding kinetics [40]. If some of the HSP90 compounds in this dataset also have time-dependent apparent IC50 values, the reported IC50s could have erroneous relative values, particularly if the kinetics vary across compounds. Another consideration is the relative solubilities of the compounds tested. A small set (30) of the 280 compounds were tested by CSAR for solubility and three were noted to have poor solubility, rendering them unmeasurable for some of the assays (e.g. ITC). While these three of the thirty HSP90 compounds were noted to have solubility issues by our CSAR colleagues, we did not have equivalent information across the dataset and we therefore treated the error estimation equivalently across the HSP90 compounds. This may or may not accurately reflect the error in the affinity measurements.
One limitation of the present study is that the modest size of the datasets, combined with the large number of scenarios that can arise in protein-ligand modeling, renders the results anecdotal in nature. Nonetheless, some of the broad conclusions appear sound and make intuitive sense, and future D3R challenges will allow these issues to be probed further and in more contexts. Moreover, the forthcoming Continuous Evaluation of Ligand Pose Predictions (CELPP: drugdesigndata.org/about/celpp) challenge will enable participants to set up and continuously evaluate their own docking servers, by using weekly pre-release data from the PDB to drive a ongoing series of blinded pose-prediction challenges. We estimate this channel will enable ~50 new pose-prediction challenges each week. A second limitation of this study is that a number of prominent research groups and companies that develop widely-used modeling tools elected not to participate. This may reduce the likelihood that the results will lead to near-term improvements in available software. Increased participation by software developers would be welcome in future challenges, and it is perhaps worth noting in this regard that submissions may be made anonymously, as detailed on the D3R website.
Finally, the present results highlight promising directions for future development. First, there appears to be considerable potential for creation of automated software and workflows that go beyond pure docking and scoring by automatically collecting and effectively using available information, such as crystal structures and affinity data, to generate enhanced pose predictions and affinity rankings. Work in this direction may ultimately benefit from integration of key data sources, such as the PDB, ChEMBL [41], BindingDB [42], and PubChem [43], to facilitate identification and collection of needed data in suitable formats. At the same time, even when prior data are available and well utilized, there is clearly also a need for improved physical models, given the difficulty of affinity calculations, even when ligand poses are known, and even when detailed simulation methods are employed. We anticipate that future developments along these and other lines will lead to continual improvement in performance on this series of blinded prediction challenges, and ultimately in the power of CADD tools to speed the design of new medications.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIH) for grant 1U01GM111528 for the Drug Design Data Resource (D3R) and U01 GM086873 to the Community Structure Activity Resource (CSAR). We are grateful to Dr. Seth Harris and to Genentech, Inc. for their collaboration and generous donation of the MAP4K4 dataset and Dr. Phil Hadjuk of Abbvie for the HSP90 dataset. We also thank OpenEye Scientific Software for generously donating the use of their software and Jenny Chong for her assistance in preparation of figures for the manuscript. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. MKG has an equity interest in, and is a cofounder and scientific advisor of, VeraChem LLC. REA has equity interest in and is a co-Founder of Actavalon, Inc., VAF has equity interest in Actavalon, Inc.
References
- 1.Smith RD, Dunbar JB, Jr, Ung PM, et al. CSAR benchmark exercise of 2010: combined evaluation across all submitted scoring functions. J Chem Inf Model. 2011;51:2115–2131. doi: 10.1021/ci200269q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Damm-Ganamet KL, Smith RD, Dunbar JB, Jr, et al. CSAR benchmark exercise 2011–2012: evaluation of results from docking and relative ranking of blinded congeneric series. J Chem Inf Model. 2013;53:1853–1870. doi: 10.1021/ci400025f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith RD, Damm-Ganamet KL, Dunbar JB, Jr, et al. CSAR Benchmark Exercise 2013: Evaluation of Results from a Combined Computational Protein Design, Docking, and Scoring/Ranking Challenge. J Chem Inf Model. 2016;56:1022–1031. doi: 10.1021/acs.jcim.5b00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlson HA, Smith RD, Damm-Ganamet KL, et al. CSAR 2014: A Benchmark Exercise Using Unpublished Data from Pharma. J Chem Inf Model. 2016;56:1063–1077. doi: 10.1021/acs.jcim.5b00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Curr Top Med Chem. 2006;6:1205–1214. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- 6.Travers J, Sharp S, Workman P. HSP90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today. 2012;17:242–252. doi: 10.1016/j.drudis.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 7.Crawford TD, Ndubaku CO, Chen H, et al. Discovery of selective 4-Amino-pyridopyrimidine inhibitors of MAP4K4 using fragment-based lead identification and optimization. J Med Chem. 2014;57:3484–3493. doi: 10.1021/jm500155b. [DOI] [PubMed] [Google Scholar]
- 8.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 9.Tembre BL, Mc Cammon JA. Ligand-receptor interactions. Comput Chem. 1984;8:281–283. doi: 10.1016/0097-8485(84)85020-2. [DOI] [Google Scholar]
- 10.Ndubaku CO, Crawford TD, Chen H, et al. Structure-Based Design of GNE-495, a Potent and Selective MAP4K4 Inhibitor with Efficacy in Retinal Angiogenesis. ACS Med Chem Lett. 2015;6:913–8. doi: 10.1021/acsmedchemlett.5b00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder P, Forster T, Kleine S, et al. Neuritogenic Militarinone-Inspired 4-Hydroxypyridones Target the Stress Pathway Kinase MAP4K4. AngewChemIntEdEngl. 2015;54:12398–12403. doi: 10.1002/anie.201501515. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Stanley M, Boggs J, et al. Fragment-based identification and optimization of a class of potent pyrrolo[2,1-f][1,2,4]triazine MAP4K4 inhibitors. BioorgMedChemLett. 2014;24:4546–4552. doi: 10.1016/j.bmcl.2014.07.071. [DOI] [PubMed] [Google Scholar]
- 13.Guimaraes CR, Rai BK, Munchhof MJ, et al. Understanding the impact of the P-loop conformation on kinase selectivity. J Chem Inf Model. 2011;51:1199–1204. doi: 10.1021/ci200153c. [DOI] [PubMed] [Google Scholar]
- 14.Huth JR, Park C, Petros AM, et al. Discovery and design of novel HSP90 inhibitors using multiple fragment-based design strategies. Chem Biol Drug Des. 2007;70:1–12. doi: 10.1111/j.1747-0285.2007.00535.x. [DOI] [PubMed] [Google Scholar]
- 15.Dunbar JB, Jr, Smith RD, Damm-Ganamet KL, et al. CSAR data set release 2012: ligands, affinities, complexes, and docking decoys. J Chem Inf Model. 2013;53:1842–1852. doi: 10.1021/ci4000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruncko M, Tahir SK, Song X, et al. N-aryl-benzimidazolones as novel small molecule HSP90 inhibitors. Bioorg Med Chem Lett. 2010;20:7503–7506. doi: 10.1016/j.bmcl.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Stanley M, Boggs JW, et al. Fragment-based identification and optimization of a class of potent pyrrolo[2,1-f][1,2,4]triazine MAP4K4 inhibitors. Bioorg Med Chem Lett. 2014;24:4546–52. doi: 10.1016/j.bmcl.2014.07.071. [DOI] [PubMed] [Google Scholar]
- 18.Molecular Operating Environment (MOE) 2016 [Google Scholar]
- 19.Read RJ, Adams PD, Arendall WB, 3rd, et al. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19:1395–1412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H, Peisach E, Westbrook JD, et al. DCC : a Swiss army knife for structure factor analysis and validation. J Appl Crystallogr. 2016;49:1081–1084. doi: 10.1107/S1600576716004428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tembre BL, Mc Cammon JA. Ligand-receptor interactions. Comput Chem. 1984;8:281–283. http://dx.doi.org/10.1016/0097-8485(84)85020-2. [Google Scholar]
- 22.OpenEye Python Toolkits 2015 [Google Scholar]
- 23.Mobley DL, Liu S, Lim NM, et al. Blind prediction of HIV integrase binding from the SAMPL4 challenge. J Comput Aided Mol Des. 2014;28:327–345. doi: 10.1007/s10822-014-9723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown SP, Muchmore SW, Hajduk PJ. Healthy skepticism: assessing realistic model performance. Drug Discov Today. 2009;14:420–427. doi: 10.1016/j.drudis.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Antosiewicz J, McCammon JA, Gilson MK. Prediction of Ph-dependent Properties of Proteins. J Mol Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. http://dx.doi.org/10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 26.Klopman G, Li J-Y, Wang S, Dimayuga M. Computer Automated log P Calculations Based on an Extended Group Contribution Approach 2002 [Google Scholar]
- 27.Ekins S, Bunin BA. The Collaborative Drug Discovery (CDD) database. Methods Mol Biol. 2013;993:139–154. doi: 10.1007/978-1-62703-342-8_10. [DOI] [PubMed] [Google Scholar]
- 28.Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Muddana HS, Fenley AT, Mobley DL, Gilson MK. The SAMPL4 host-guest blind prediction challenge: an overview. J Comput Aided Mol Des. 2014;28:305–317. doi: 10.1007/s10822-014-9735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papadatos G, Gaulton A, Hersey A, Overington JP. Activity, assay and target data curation and quality in the ChEMBL database. J Comput Aided Mol Des. 2015;29:885–896. doi: 10.1007/s10822-015-9860-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawkins PC, Skillman AG, Warren GL, et al. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar A, Zhang KYJ. A pose prediction approach based on ligand 3D shape similarity. J Comput Aided Mol Des. 2016;30:457–69. doi: 10.1007/s10822-016-9923-2. [DOI] [PubMed] [Google Scholar]
- 33.Kumar A, Zhang KYJ. Prospective evaluation of shape similarity based pose prediction method in D3R Grand Challenge 2015. J Comput Aided Mol Des. 2016 doi: 10.1007/s10822-016-9931-2. [DOI] [PubMed] [Google Scholar]
- 34.Rush TS, 3rd, Grant JA, Mosyak L, Nicholls A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction. J Med Chem. 2005;48:1489–1495. doi: 10.1021/jm040163o. [DOI] [PubMed] [Google Scholar]
- 35.Cozzetto D, Tramontano A. Relationship between multiple sequence alignments and quality of protein comparative models. Proteins. 2005;58:151–157. doi: 10.1002/prot.20284. [DOI] [PubMed] [Google Scholar]
- 36.Kelley BP, Brown SP, Warren GL, Muchmore SW. POSIT: Flexible Shape-Guided Docking For Pose Prediction. 2015 doi: 10.1021/acs.jcim.5b00142. [DOI] [PubMed] [Google Scholar]
- 37.Kim R, Skolnick J. Assessment of programs for ligand binding affinity prediction. J Comput Chem. 2008;29:1316–1331. doi: 10.1002/jcc.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gumbart JC, Roux B, Chipot C. Efficient Determination of Protein–Protein Standard Binding Free Energies from First Principles. J Chem Theory Comput. 2013;9:3789–3798. doi: 10.1021/ct400273t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gumbart JC, Roux B, Chipot C. Standard Binding Free Energies from Computer Simulations: What Is the Best Strategy? J Chem Theory Comput. 2013;9:794–802. doi: 10.1021/ct3008099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gooljarsingh LT, Fernandes C, Yan K, et al. A biochemical rationale for the anticancer effects of Hsp90 inhibitors: Slow, tight binding inhibition by geldanamycin and its analogues. Proc Natl Acad Sci. 2006;103:7625–7630. doi: 10.1073/pnas.0602650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaulton A, Bellis LJ, Bento AP, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–7. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu T, Lin Y, Wen X, et al. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim S, Thiessen PA, Bolton EE, et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016;44:D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.