

Summary
Naturally occurring, pharmacologically active peptides constrained with covalent crosslinks generally have shapes evolved to fit precisely into binding pockets on their targets. Such peptides can have excellent pharmaceutical properties, combining the stability and tissue penetration of small molecule drugs with the specificity of much larger protein therapeutics. The ability to design constrained peptides with precisely specified tertiary structures would enable the design of shape-complementary inhibitors of arbitrary targets. Here we describe the development of computational methods for de novo design of conformationally-restricted peptides, and the use of these methods to design 15–50 residue disulfide-crosslinked and heterochiral N-C backbone-cyclized peptides. These peptides are exceptionally stable to thermal and chemical denaturation, and twelve experimentally-determined X-ray and NMR structures are nearly identical to the computational models. The computational design methods and stable scaffolds presented here provide the basis for development of a new generation of peptide-based drugs.
Main Text
The vast majority of drugs currently approved for use in humans are either proteins or small molecules. Lying between the two in size, and integrating the advantages of both1,2, constrained peptides are an underexplored frontier for drug discovery. Naturally-occurring constrained peptides, such as conotoxins, chlorotoxin, knottins, and cyclotides, play critical roles in signaling, virulence and immunity, and are among the most potent pharmacologically active compounds known3. These peptides are constrained by disulfide bonds or backbone cyclization to favour binding-competent conformations that precisely complement their targets. Inspired by the potency of these compounds, there have been considerable efforts to generate new bioactive molecules by re-engineering existing constrained peptides using loop grafting, sequence randomization, and selection4. Although powerful, these approaches are hindered by the limited variety of naturally-occurring constrained peptide structures and the inability to achieve global shape complementarity with targets. There is need for a method of creating constrained peptides with new structures and functions that provides precise control over the size and shape of the designed molecules. A method with sufficient generality to incorporate noncanonical backbones and unnatural amino acids would enable access to broad regions of peptide structure and function space not explored by evolution.
Although there have been recent advances in protein design methodology5–9, the computational design of covalently-constrained peptides with new structures and noncanonical backbones presents new challenges. First, both backbone generation and design validation by structure prediction require new backbone sampling methods which can handle cyclic and mixed-chirality backbones. Second, methods are needed for incorporation of multiple covalent geometric constraints without introduction of conformational strain. Third, energy evaluations must correctly model amino acid chirality.
Here we describe the development of new computational methods that meet these challenges, opening this exciting frontier to computational design. We demonstrate the power of the methods by designing a structurally diverse array of 15–50 residue peptides spanning two broad categories: (i) genetically-encodable disulfide-rich peptides, and (ii) heterochiral peptides with noncanonical architectures and sequences. Genetic encodability has the advantage of being compatible with high-throughput selection methods, such as phage, ribosome, and yeast display, while incorporation of noncanonical components allows access to new types of structures, and can confer enhanced pharmacokinetic properties. To explore the folds accessible to genetically-encoded constrained peptides under 50 amino acids, we selected nine topologies: HH, HHH, EHE, EEH, HEEE, EHEE, EEHE, EEEH, and EEEEEE (Fig. 1; we define a “topology” as the sequence of secondary structure elements in the folded peptide, where H denotes α-helix and E denotes β-strand). To explore the expanded design space accessible with inclusion of noncanonical amino acids and backbone cyclization, we sought to cover all topologies containing two to three canonical secondary structure elements: HH, HHH, EEH, EHE, HEE, and EE, along with HLHR, a cyclic topology with right- and left-handed helices.
Figure 1. Designed peptide topologies.
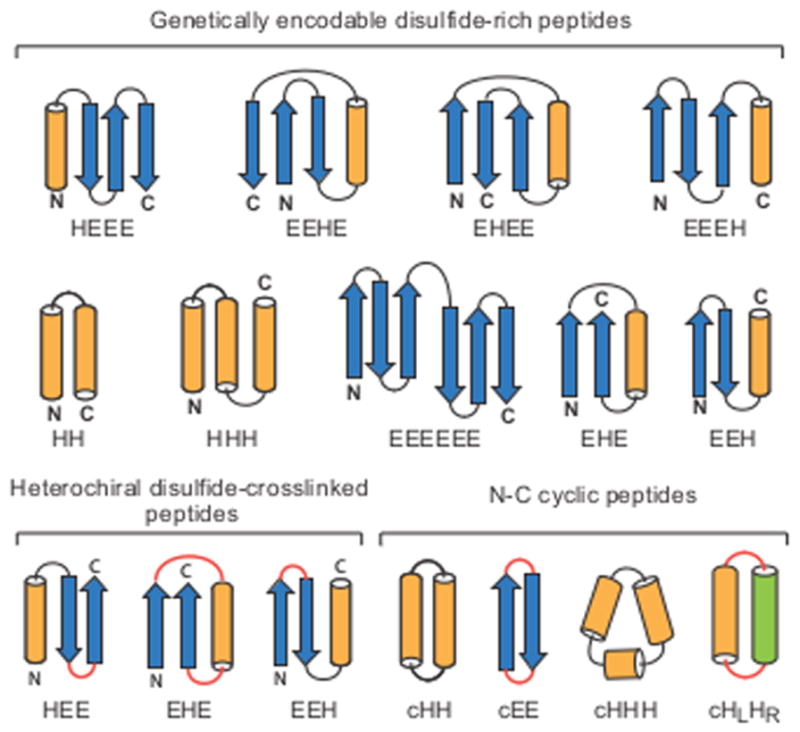
The designed secondary structure architectures for each of the three classes of constrained peptides (genetically-encodable disulfide-rich, heterochiral disulfide-crosslinked, and cyclic) span most of the topologies that can be formed with four or fewer secondary structure elements. Arrows: β-strands, orange cylinders: right-handed α-helices, green cylinder: left-handed α-helix; red: loop segments containing D-amino acid residues.
All of the design calculations described in this paper were carried out with the Rosetta software suite10 and followed the same basic approach. Large numbers of peptide backbones were stochastically generated as described in the following sections, combinatorial sequence design calculations were carried out to identify sequences (including disulfide crosslinks) stabilizing each backbone conformation, and the designed sequence-structure pairs were assessed by determining the energy gap between the designed structure and alternative structures found in large-scale structure prediction calculations based on the designed sequence. A subset of the designs in deep energy minima were then produced in the laboratory, and their stabilities and structures were determined experimentally.
Genetically-encodable disulfide-constrained peptides
To design disulfide-stabilized genetically-encodable peptides, we created a “blueprint” specifying the lengths of each secondary structure and connecting loop for each topology. Ensembles of backbone conformations were generated for each blueprint by Monte Carlo-based assembly of short protein fragments9, or, in the case of HH and HHH topologies, by varying the parameters in parametric generating equations11. The backbones were scanned for sites capable of hosting disulfide bonds with near-ideal geometry, and one to three disulfide bonds were incorporated. Low-energy amino acid sequences were designed for each disulfide-crosslinked backbone using iterative rounds of Monte Carlo-based combinatorial sequence optimization while allowing the backbone and disulfide linkages to relax in the Rosetta all-atom force field (see Methods). Except for the EHEE topology, we performed no manual amino acid sequence optimization. Rosetta ab initio structure prediction calculations were carried for each designed sequence, and synthetic genes were obtained for a diverse set of 130 for which the target structure was in a deep global free energy minimum (Fig. 2a,b).
Figure 2. Computational design and biophysical characterization of genetically-encodable disulfide-rich peptides.

Genetically-encodable peptides are given the prefix “g” and a number to differentiate designs that share a common topology. (column a) Cartoon renderings of each design are shown with rainbow colouring from the N-terminus (blue) to the C-terminus (red), and disulfide bonds are shown as sticks. (column b) The energy landscape of each designed sequence was assessed by Rosetta structure prediction calculations starting from an extended chain (blue dots) or from the design model (orange dots); lower energy structures were sometimes sampled in the former because disulfide constraints were only present in the latter. (column c) CD spectra at 20°C (blue line), after heating to 95°C (red line), and upon cooling back to 20°C (green line). Spectra collected with 2.5 mM TCEP are shown in purple. (column d) CD steady-state wavelength spectra as a function of GdnHCl concentration.
Disulfide bonds in peptides are unlikely to form in the reducing environment of the cytoplasm, so designs were secreted from Escherichia coli or cultured mammalian cells12 (see Methods). Twenty-nine designs exhibited a redox-sensitive gel-shift, redox-sensitive HPLC migration, and/or a CD spectrum consistent with the designed topology (see Supplementary Document 3). All twenty-nine contain at least one non-alanine hydrophobic residue on each secondary structure element contributing van der Waals interactions in the core, which are likely important for proper peptide folding. We chose one representative design from each topology for further biochemical characterization. Since eight of the nine topologies contained four or more cysteine residues, we used multiple-stage mass spectrometry to investigate the disulfide connectivity. In all cases the data were consistent with the designed connectivity (see Supplementary Document 4).
The stability of the designs to thermal and chemical denaturation was assessed by CD spectroscopy. Samples were heated to 95°C (Fig. 2d), or incubated with increasing concentrations of guanidinium hydrochloride (GdnHCl) (Fig. 2e). The contribution of disulfide bonds to protein folding was assessed by incubating samples with a ~100-fold molar excess of the reductant tris(2-carboxyethyl)phosphine (TCEP). Designs gHEEE_02, gEEEH_04, and gEEEEEE_02 are resistant to both thermal and chemical denaturation, while design gHH_44 is resistant to thermal denaturation. gHEEE_02 contains three disulfide bonds, with each secondary structure element participating in at least one disulfide bond, and no two secondary structure elements sharing more than one disulfide bond. gEEEH_04 has two of three disulfide bonds linking the N-terminal β-strand to the C-terminal α-helix. gEEEEEE_02 consists of two antiparallel β-sheets packing against one another in a sandwich-like arrangement, with each β-sheet stabilized by a disulfide bond linking one terminus to its adjacent β-strand. gHH_44 consists of two α-helices with a single disulfide bond connecting the termini.
We crystallized design gEHEE_06 and determined the structure to a resolution of 2.09 Å (Fig. 3, Supplementary Information Table S2-2). The crystals had threefold non-crystallographic symmetry, and each protomer aligns to the design model with a mean all-atom RMSD of 1.12 Å. All three of the designed disulfide bonds were well-defined by electron density (Extended Data Fig. 1), and rotamers of core residues exhibited excellent agreement with the design model. The protein was thermostable and completely resistant to chemical denaturation (Fig. 2d,e). While gEHEE_06 shares the short-chain scorpion toxin topology, the length of secondary structure elements and loops, and the position of the disulfide bonds, are entirely divergent from known natural peptides.
Figure 3. X-ray crystal structures and NMR solution structures of designed peptides are very close to design models.

Structures for gEHE_06, gEEH_04, gEEHE_02, and gHHH_06 were determined by NMR spectroscopy, and the structure of gEHEE_06 was determined by X-ray crystallography. (column a) Cα traces of NMR ensembles, or superimposed members of the asymmetric unit, (grey) are aligned against the design model (rainbow). Disulfide bonds are shown with sidechain atoms rendered as sticks with sulfur atoms coloured yellow. (column b) A cartoon representation of the lowest energy conformer of each NMR ensemble or crystallographic asymmetric unit (grey) is shown aligned to the design model (rainbow). Sidechain atoms of hydrophobic core residues are rendered as sticks.
As crystallization efforts for other designs were unsuccessful (with phase-separation rather than protein precipitation observed), we expressed isotope-labelled peptides in E. coli, and determined structures by nuclear magnetic resonance (NMR) spectroscopy13,14 (see Methods). Upfield chemical shifts of the cysteine β-carbons15 (deposited in the BMRB) confirmed the formation of the designed disulfide bonds. Design gEEHE_02, with one disulfide bond connecting the termini within the β-sheet and two between the α-helix and β-sheet, aligns to the NMR ensemble with a mean all-atom RMSD of 1.44 Å. This design was impervious to both thermal and chemical denaturation (monitored by CD spectroscopy), and remained partially folded in the presence of TCEP. The final three designs are each composed of three secondary structure elements, with termini located at opposite ends of the molecule and two disulfide bonds connecting each terminus to the middle structural element or adjacent loop. gEEH_04 was less stable than the others to thermal denaturation, but its NMR structure is nearly identical to the design model (mean all-atom RMSD of 1.29 Å). gEHE_06, which contains a solvent-exposed two-strand parallel β-sheet (rare in natural protein structures16), aligns to the NMR ensemble with an all-atom mean RMSD of 1.95 Å. It was thermally and chemically stable based on CD measurements, and remained folded in the presence of TCEP. gHHH_06 partially unfolds upon heating to 95°C but returns to the folded state upon cooling; the design model aligns to the NMR ensemble with a mean all-atom RMSD of 1.74 Å. Taken together, the X-ray crystallographic and NMR structures demonstrate that our computational approach enables accurate design of protein mainchain conformation, disulfide bonds, and core residue rotamers.
Synthetic heterochiral disulfide-constrained peptides
We next sought to design shorter disulfide-constrained peptides incorporating both L- and D-amino acids. We generalized the Rosetta energy function to support D-amino acids by inverting the torsional potentials used for the equivalent L-amino acids (see Methods and Supplementary Information), and sequence design algorithms were extended to enable mixed-chirality design. Since chemical synthesis is labour-intensive, we prioritized the development of automated computational screening techniques, supplementing Rosetta ab initio screening with molecular dynamics (MD) evaluation.
Large numbers of disulfide-constrained backbones for topologies HEE, EHE, and EEH were generated by fragment assembly as described above for genetically-encodable peptides. Sequences were designed (permitting D-amino acids at positive-phi positions), and the resultant low-energy designs were evaluated using MD and ab initio structure prediction (Extended Data Fig. 2). For each topology, we selected a single, low-energy design (Extended Data Fig. 3) which underwent only small (< 1.0 Å RMSD) fluctuations in the MD simulations (Extended Data Fig. 4) and had a significant energy gap in the structure prediction calculations. Selected peptides were chemically synthesized, and structurally characterized by NMR. In all three cases, the NMR spectra had well-dispersed, sharp peaks and secondary 1Hα chemical shifts consistent with the secondary structure of the design model (Supplementary Fig. S2-5).
High-resolution NMR solution structures were determined for each of the designs (Supplementary Information Table S2–3). NC_HEE_D1 is a 27-residue peptide with a D-proline, L-proline turn at the β-β junction; in this case, Rosetta re-identified a motif known previously to stabilize type II′ turns17,18. The NMR structure closely matches the design model: the Cα RMSD is 0.99 Å between the designed structure and the lowest-energy NMR model (Fig. 4, top row). NC_EHE_D1 is a 26-residue peptide crosslinked using two disulfide bonds with a D-arginine residue in the β-α loop and a D-asparagine residue as the C-terminal capping residue for the α-helix. The design model has a 1.9 Å Cα RMSD to the lowest-energy NMR ensemble member, and 0.68 Å Cα RMSD to the closest member of the ensemble (Fig. 4, middle row; the last two residues at C-terminal vary considerably in the ensemble). NMR characterization of NC_EEH_D1 design showed an unwound C-terminal α-helix adopting an extended conformation, differing from the design model (Extended Data Fig. 5). We hypothesized that substantial strain was introduced by the angle between the helix and the preceding strand, and by the disulfide bonds at both ends of the helix. A second design for the same topology, NC_EEH_D2, has a type I′ turn at the β-β connection and a different disulfide pattern. The NMR ensemble for NC_EEH_D2 is very close to the design model (0.86 Å Cα RMSD to the lowest-energy NMR model; Fig. 4, bottom row).
Figure 4. Design and characterization of heterochiral disulfide-constrained peptides.

The prefix “NC” denotes noncanonical sequence or backbone architecture, and a numerical suffix differentiates designs sharing a common topology. (Column a) Cartoon representations of design models with the N-terminus in blue and C-terminus in red. (Column b) Folding energy landscapes from Rosetta ab initio structure prediction calculations. Blue dots indicate lowest-energy structures identified in independent Monte Carlo trajectories. Orange dots are from trajectories starting with the design model. (r.e.u: Rosetta Energy Units, RMSD: root mean square deviation from the designed topology). (Column c) Five representative trajectories from a total of 50 independent molecular dynamics simulations starting from the design model with different initial velocities. (Column d) NMR-determined structure ensembles. Cartoon representations coloured and oriented as in column a. (Column e) Superposition of the designed structure (blue) with the lowest-energy NMR structure (green). (Column f) CD wavelength spectra between 195 nm and 260 nm recorded at 25 °C (black), 55 °C (blue), 95 °C (red), and after cooling back to 25 °C (green). (Column g) CD spectra recorded at 0 M (black), 2 M (blue), 4 M (green), or 6 M GdnHCl (red), or with 2.5 mM TCEP/0 M GdnHCl (purple). Data are truncated in the far-UV region for spectra acquired in the presence of high GdnHCl concentrations (due to GdnHCl absorbance).
We explored the stability of the designed peptides using CD spectroscopy to monitor thermal and chemical denaturation. All three peptides are very thermostable: there is no loss in secondary structure for NC_HEE_D1 and NC_EEH_D2 at 95 °C, and only a small decrease for NC_EHE_D1 (Fig. 4f). Remarkably, NC_HEE_D1 does not denature in 6 M GdnHCl (Fig. 4g, top row). Treatment with TCEP causes unfolding of all three designs, highlighting the importance of disulfide bonds.
Both the genetically-encoded and noncanonical disulfide crosslinked designs were created de novo without sequence information from natural proteins. Searches for similar sequences in the PDB and NCBI non-redundant database using PSI-BLAST found a significant alignment (e-value < 0.01) only for NC_EHE_D1. This sequence has weak similarity (e-value of 2×10−4) to the zinc-finger domain of lysine-specific demethylase (PDB ID: 2MA5), but the aligned regions adopt different structures (Extended Data Fig. 6).
Synthetic backbone-cyclized peptides
Next, we explored the design of peptides with cyclized backbones, which can increase stability and protect against exopeptidases19. To generate such backbones without dependence on fragments of known structures, we implemented a generalized kinematic loop closure20,21 method (named “GenKIC”) to sample arbitrary covalently-linked atom chains capable of connecting the termini. Each GenKIC chain-closure attempt involves perturbing multiple chain degrees of freedom, then analytically solving kinematic equations to enforce loop closure with ideal peptide bond geometry in the case of N-C cyclic peptides (see Methods, Supplementary Information, and Extended Data Fig. 7). Sequence design, backbone relaxation, and in silico structure validation using MD simulation and Rosetta ab initio structure prediction were carried out with terminal bond geometry constraints (Extended Data Fig. 2).
We synthesized cyclic peptides for three topologies (cEE, cHH, and cHHH) and their structures were determined by NMR spectroscopy. The 18-residue NC_cEE_D1 design has a cyclic anti-parallel β-sheet fold similar to natural theta-defensins, but with one disulfide bond (rather than three), and different turn types containing heterochiral sequences22. The lowest-energy NMR model has a Cα RMSD of 1.26 Å to the designed structure. The variability in the curvature of the sheets across the NMR ensemble is similar to the variability observed in the structure calculations (Fig. 5, top row). The 26-residue NC_cHH_D1 design, which has one disulfide bond linking the two α-helices, has a 1.03 Å Cα RMSD from the lowest-energy NMR structure (Fig. 5, second row). The 22-residue NC_cHHH_D1 design has three short regions of α-helical structure and a single disulfide bond. The NMR structure of the design was again very close to the design model (Fig. 5, third row), with a Cα RMSD of 1.06 Å to the lowest-energy NMR structure.
Figure 5. Design and characterization of N-C backbone cyclic peptides.

Columns are as indicated in Figure 4 legend. A lowercase “c” in the peptide name indicates N-C cyclic backbone.
All three cyclic topologies were found to be extremely stable in thermal denaturation experiments, retaining CD signal when heated to 95 °C (Fig. 5f). The CD spectra of NC_cHH_D1 and NC_cEE_D1 were nearly identical in 0 and 6 M GdnHCl, indicating that these peptides do not chemically denature (Fig. 5g; NC_cHHH_D1 showed some loss of secondary structure in 6M GdnHCl). After treatment with TCEP, both NC_cHH_D1 and NC_cHHH_D1 lost secondary structure, but the CD spectrum of NC_cEE_D1 was not changed by reduction of the central disulfide bond (Fig. 5g, top row). Overall, the cyclic designs are exceptionally stable given their very small sizes.
Beyond natural secondary and tertiary structure
As a final test of the generality of the new design methodology, we designed a heterochiral, backbone-cyclized, two-helix topology with one noncanonical left-handed α-helix and one canonical right-handed α-helix (HLHR) assembling into a tertiary structure not observed in natural proteins. As before, we validated designs by MD; however, for validation by ab initio structure prediction it was necessary to develop a new, GenKIC-based structure prediction protocol (see Extended Data Fig. 8, Methods, and Supplementary Information) since the standard Rosetta ab initio structure prediction method utilizes fragments of native proteins, which typically do not contain left-handed helices. Our selected design for this topology, NC_HLHR_D1, is a 26-residue peptide with one D-cysteine, L-cysteine disulfide bond connecting the right-handed and left-handed α-helices. There is an excellent match between the NMR structure ensemble and design model (Cα RMSD: 0.79 Å) (Fig. 6). As expected for the nearly achiral topology, the CD signal is very small (as observed for a previously-studied two-chain, four-helix mixed D/L system23), and no change was observable on heating to 95 °C. The secondary 1Hα chemical shifts also show no significant change on heating to 75 °C (Fig. 6g, Supplementary Fig. 2–6), indicating that the peptide is thermostable. Successful design of this topology demonstrates that our computational methods are sufficiently versatile and robust to design in a conformational space not explored by nature.
Figure 6. Design and characterization of a peptide with noncanonical secondary and tertiary structure.

a) NC_HLHR_D1 design (cyan: L-amino acids, orange: D-amino acids) b) Folding energy landscape generated using a new structure prediction algorithm compatible with noncanonical secondary structures (see Methods and Supplementary Information). c) Five representative molecular dynamics trajectories (from a total of 50) starting from the design model with different initial velocities. d) NMR-determined structure ensembles, coloured and oriented as in first panel. e) Superposition of designed structure (blue) with lowest-energy NMR structure (green). f) CD spectra between 195 nm and 260 nm recorded at 25 °C (black), 55 °C (blue), 95 °C (red), and after cooling back to 25 °C (green). The CD spectrum of NC_HLHR_D1 exhibits very weak signals because the L- and D- helical signals largely cancel. g) Secondary 1Hα chemical shifts (ppm) show no change from 25 °C (black) to 75 °C (red).
Conclusions
The key advances in computational design presented here — notably the methods for designing constrained peptide backbones spanning a broad range of topologies and incorporating natural and non-natural building-blocks — enable high-accuracy design of new peptides with exceptional thermostability and resistance to chemical denaturation. All twelve experimentally-determined structures are in close agreement with the design models, including one with helices of different chirality. Unlike the natural constrained peptide families, designed peptides are not limited to particular shapes, sizes, nucleating motifs, or disulfide connectivities; indeed, the sequences of these de novo peptides are quite different from those of any known peptides. In this paper, we have focused on extending sampling and scoring methods to permit design with D-amino acids and cyclic backbones, but the new tools are fully generalizable to peptides containing more exotic building-blocks, such as amino acids with noncanonical sidechains24 or noncanonical backbones25.
The hyperstable molecules presented in this study provide robust starting scaffolds for generating peptides that bind targets of interest using computational interface design26 or experimental selection methods. Solvent-exposed hydrophobic residues can be introduced without impairing folding or solubility (Extended Data Figs. 9 and 10, Supplementary Fig. S2–6) suggesting high mutational tolerance. Hence it should be possible to reengineer the peptide surfaces, incorporating target-binding residues to construct binders, agonists, or inhibitors. There has been considerable effort in both academia and industry to employ small, naturally-occurring proteins as alternatives to antibody scaffolds for library selection-based affinity reagent generation. Our genetically-encoded designs offer considerable advantages as starting points for such approaches because of their high stability, small size, and diverse shapes. Furthermore, having been designed exclusively to be robust and stable, they lack the often-destabilizing non-ideal structural features that arise in naturally occurring proteins from evolutionary selective pressure for a particular function. Similarly, the heterochiral designs described here provide starting points for split-pool and other selection strategies compatible with noncanonical amino acids.
Going beyond the reengineering of our hyperstable designs to bind targets of interest, the methods developed in this paper can be used to design new backbones to fit specifically into target binding pockets. Such “on-demand” target-specific scaffold generation is likely to yield scaffolds with considerably greater shape-complementarity than that of scaffolds generated without knowledge of the target. More generally, our computational methods open up previously inaccessible regions of shape space, and, in combination with computational interface design, should help unlock the pharmacological potential of peptide-based therapeutics.
Methods
Computational design
De novo design of constrained peptides can be divided into two main steps: backbone assembly and sequence design. Practically, our peptide design pipeline has been optimized to permit these two steps to be performed in immediate succession with a single set of inputs, with no need for export or manual curation of generated backbones prior to the sequence design. (A third and final validation step is typically performed separately.)
For backbone assembly, we used two different approaches in this report: disulfide-constrained topologies were sampled using a fragment assembly method, while backbone-cyclized peptide topologies were sampled using a fragment-independent kinematic closure-driven approach. Example scripts and command lines for each step in the design workflow are available in the Supplementary Information.
Backbone design using fragment assembly
In the case of disulfide-crosslinked designs, the topology was defined using a “blueprint” that specifies secondary structure and torsion bins for each amino acid residue, the latter defined using the ABEGO alphabet system described previously7,9. The ABEGO nomenclature assigns a letter to each of five regions, or bins, in Ramachandran space. These correspond to the α-helical region (A), the β-sheet region (B), the region with positive phi values typically accessed by glycine (G), and the remainder of the Ramachandran space (E). (The fifth bin, O, represents residues with cis-peptide bonds, and was not used here.) The blueprint is the input for a Rosetta Monte Carlo-based fragment assembly protocol7,9,10,27 that generates backbone conformations matching the blueprint architecture. Briefly, the fragment assembly protocol uses the defined blueprint to pick backbone fragments from a database of non-redundant high-resolution crystal structures. The insertion of fragments serves as the moves in a Monte Carlo search of backbone conformation space. For searches of the NC_EEH topology, loop types were limited to ABEGO bins EA and GG for the ββ connection, and BAB and GBB for the αβ connection. For sampling of the NC_EHE topology, βα connections were limited to GBB, BAB, and AB, while αβ connections were limited to GB, GBA, and AGB. For sampling of the NC_HEE topology, αβ connections were limited to BAAB, GB, GBA, and AGB, while ββ connections were limited to EA and GG.
Backbone design using generalized kinematic closure
While the fragment-based approaches described above are powerful, they are limited to conformations favored by peptides composed primarily of L-amino acids. For N-C cyclic designs — NC_cHHH_D1, NC_cHH_D1, NC_cEE_D1, NC_cHLHR_D1 — we chose to focus on fragment-independent methods that are better suited to explore conformations that are only accessible to mixed D/L peptides. We therefore turned to generalized kinematic closure (GenKIC).
GenKIC-based sampling works by treating a peptide as a loop, or series of loops, to be “closed”. The torsion values of an initial, “anchor” residue are randomly selected; this residue is then fixed, and the rest of the peptide is treated as a loop closure problem. The particular covalent linkages serve as a set of geometric constraints for loop closure. The GenKIC algorithm performs a series of user-controlled perturbations to the torsion angles of the peptide chain, which inevitably disrupt the geometry of the closure points. GenKIC then mathematically solves for the value of six “pivot” torsion angles that restore the geometry of the closure points and permit the loop to remain closed20,21,28. Since the algorithm can return up to sixteen solutions per closure attempt, a filters are applied to eliminate solutions with pivot amino acid residues in energetically unfavorable regions of Ramachandran space or with other geometric problems, such as clashes with other residues. The “best” solution is then chosen based on the Rosetta score function10.
During the sampling steps, regions in the designed topology that were intended to form helices or sheets were initialized to ideal phi/psi values, and were either kept fixed or perturbed by only small amounts (<20 degrees). In loop regions, the perturbation was carried out by drawing torsion values randomly, biased by the Ramachandran preferences of the amino acid residue. Glycine or D/L alanine were used for backbone sampling prior to design. The allowed torsion value range either covered the entire Ramachandran space, or, in cases in which known loop ABEGO patterns could connect secondary structure elements, the mainchain torsion values were were limited to those ABEGO bins. For example, during the design of the cEE topology, connection types were limited to the ‘GG’ and ‘EA’ torsion bins for the 2-residue loops.
Disulfide positioning
To design disulfide bonds, we first evaluated all residue pairs with Cβ atoms ≤ 5 Å apart for geometry suitable to disulfide bond formation27, selected backbones that could harbor disulfide bonds with near-ideal geometry, and incorporated one to three disulfide bonds. To select an ideal disulfide configuration from the set of all sterically possible combinations of disulfide bonds for a given backbone, we ranked disulfide configurations according to their effect on the unfolded state configurational entropy. The reduction in unfolded state entropy due to a set of multiple crosslinks was computed according to a random flight model using Eq. 6 in Harrison et al.29, with ΔV = 29.65 Å3 and b = 3.8 Å3. This method has been implemented in the Rosetta software suite as the Disulfidize Mover and DisulfideEntropy Filter, both of which are accessible to the RosettaScripts scripting language.
Modifications to Rosetta to permit design of cyclic backbones and mixed D/L peptides
D-amino acid residues allow access to regions of conformational space normally only accessed by glycine. When placed correctly, they can provide greater rigidity than glycine, stabilizing glycine-dependent structural motifs and, thereby, the overall fold30. Because the Rosetta software suite has primarily been used for designing proteins consisting of the 19 canonical L-amino acids and glycine, a number of modifications were necessary in order to permit robust design of peptides containing mixtures of D- and L-amino acids. First, Rosetta’s default scoring function (talaris2013 at the time of the work described here) was updated to permit D-amino acids to be scored with mirror symmetry relative their L-counterparts. Terms in the score function that are based on mainchain or sidechain torsion values were modified to invert D-amino acid torsion values before applying the equivalent L-amino acid potentials. Those score function terms that are based on interatomic distances required minimal changes. To permit energy minimization, score function derivatives were also modified to invert torsion derivative values for D-amino acids. Rosetta’s rotameric search algorithm, the packer, was modified to use L-amino acid rotamers with sidechain chi torsion values inverted for D-amino acid rotamer packing, and to update Hα and Cβ positions appropriately when inverting residue chirality. Finally, we added an option to symmetrize the energy tables for the mainchain torsion preferences of glycine, which are asymmetric by default because they are based on statistics taken from the Protein Data Bank. (Glycine, in the context of L-amino acids only, occurs disproportionately in the positive-phi region of Ramachandran space, but should have no asymmetric preferences in a mixed D/L context.) Details of these modifications are described in the Supplementary Information.
Because Rosetta has traditionally been used to build linear polymers, a number of core Rosetta libraries had to be modified to permit N-C cyclic geometry to be sampled and scored properly. The assumption that residue i is connected to residues i+1 and i−1, which is invalid for cyclic peptides, has been removed and replaced with proper lookups of connected residue indices. Cyclic geometry support was tested by confirming that the circular permutations of cyclic peptide models score identically.
Note that, as of 11 March 2016, the default Rosetta score function has been changed to talaris2014, which re-weights a number of score terms and introduces one new term31. The talaris2014 score function has also been made fully compatible with D-amino acids and cyclic geometry. A newer, experimental score function, currently called beta_nov15, has also been made fully compatible with D-amino acids and cyclic geometry.
Sequence design and filtering
Backbone assembly using fragment assembly or GenKIC was followed by a sequence design step. Sequence design was performed using the FastDesign protocol (see Supplementary Information). This involves four rounds of alternating sidechain rotamer optimization (during which sidechain identities were permitted to change) and gradient descent-based energy minimization. The best-scoring structure was taken from a minimum of three repeats of FastDesign (twelve rounds of rotamer optimization and minimization). Each amino acid position was sorted into a layer (“core”, “boundary”, or “surface”) based on burial, and the layer dictated the possible amino acid types allowed at that position. Hydrophobic amino acid residues, for example, were only permitted at core positions. To favor more proline residues during sequence design, the reference weight for proline in the Rosetta score function was reduced by 0.5 units. Backbones were allowed to move during the relaxation steps. For each topology ~80,000 structures were generated, and filtered based on the overall energy per residue, score terms related to backbone quality, and score terms related to the disulfide geometry. In a few cases for noncanonical peptides, a conservative mutation was manually introduced into a surface-exposed repeat sequence (e.g. an arginine to break a poly-lysine sequence) to facilitate unambiguous NMR assignment.
Rosetta-based computational validation
Typically, the number of designs that can be created in silico exceeds the number that can be produced and examined experimentally. We therefore used Rosetta to prune the list of designs, by one of two methods. For design consisting of canonical amino acids, Rosetta’s fragment-based ab initio algorithm32 was utilized to predict a design’s structure given its amino acid sequence, and to determine whether the target structure was a unique minimum in the conformational energy landscape. Disulfide bonds were not allowed to form during these simulations; the designed disulfide bonds are intended to stabilize the folded conformation rather than direct protein folding. Designs which incorporate short stretches of D-amino acids were also validated using Rosetta’s fragment-based ab initio algorithm; the amino acid sequences of designs, with all D-amino acids mutated to glycine, were provided as input, and we allowed Rosetta to generate on the order of 30,000 predicted structures as output. Unlike the standard ab initio protocol, we did not use secondary structure predictions in fragment picking. Additionally, the length of small and large fragments was set to 4 and 6 amino acid residues, instead of the default 3 and 9; we found that this produced better sampling for peptides. After conformational sampling, the D-amino acid positions were changed to their original identities, and rescored. A small modification to the ab initio algorithm permitted it to build a terminal peptide bond for the N-C cyclic designs during the full-atom refinement stages of the structure prediction. Those designs that showed no sampling near the design conformation, or for which the design conformation was not the unique, lowest-energy conformation, were discarded.
Since fragment-based methods are poorly suited to the prediction of structures with large amounts of D-amino acid content, such as NC_cHLHR_D1, we developed a new, fragment-free algorithm for validation of these topologies. This algorithm, which we call “simple_cycpep_predict”, uses the same GenKIC-based sampling approach used to build backbones for design, with additional steps of filtering solutions based on disulfide geometry, optimizing sidechain rotamers, and gradient-descent energy minimization. Because the search space is vast, even with the constraints imposed by the N-C cyclic geometry and the disulfide bond(s), we further biased the search by setting mainchain torsion values for residues in the middle of the helices to helical values (a Gaussian distribution centred on phi=−61°, psi=−41° for the αR helix and on phi=+61°, psi=+41° for the αL helix); this is analogous to the biased sampling obtained by fragment-based methods, in which sequences with high helix propensity are sampled primarily with helical fragments. As with ab initio validation, designs showing poor sampling near the design conformation or poor energy landscapes were discarded.
Molecular dynamics-based computational validation
We carried out further molecular dynamics-based validation of those designs for which the ab initio or simple_cycpep_predict algorithms predicted high-quality energy landscapes. Similar to strategies described previously33,34, we used multiple short and independent trajectories, starting with different initial velocities to analyze the conformational flexibility and kinetic stability of designed peptides. MD simulations were performed in explicit solvent conditions using the AMBER12 package and Amber ff12sb force field35. A rectangular water box with 10 Å buffer of TIP3P water36 in each direction from the peptide was used for simulations. Sodium and chloride counterions were added to neutralize the system. The solvated system was minimized in two steps: solvent was first minimized for 20,000 cycles while keeping restraints on the peptide, followed by minimization of the whole system for another 20,000 cycles. At the start of simulations, the system was slowly heated from 0 K to 300 K under constant volume with positional restraints on the peptide of 10 kcal/(mol·Å) for 0.1 ns. For each selected peptide, 50 independent simulations starting with different initial velocities were performed. Each simulation started with the energy-minimized designed model, and was carried out for ~3.5 ns. Periodic boundary conditions were used with a constant temperature of 300 K using the Langevin thermostat37 and a pressure of 1 atm with isotropic molecule-based scaling. A cutoff of 10 Å was used for the Lennard-Jones potential and the Particle Mesh Ewald method38 to calculate long-range electrostatic interactions. The SHAKE algorithm39 was applied to all bonds involving H atoms and an integration step of 2 fs was used for the simulations with amber12 PMEMD in the NPT ensemble. At the conclusion of the simulations, all the trajectories were analysed using the Amber12 package and VMD40. We looked for for fluctuations in RMSD, and for the convergence (or the lack thereof) to the designed structure among all the trajectories. Distribution of RMSD values at the end of all trajectories was also analyzed, although the beginning two-thirds of each trajectory were discarded as a burn-in period. MD analyses for three designs of the same topology are shown in Extended Data Figure 4.
Prediction of mutational tolerance
Since the designed peptides presented in this study are intended to be used as starting points for designing binders to targets of therapeutic interest, we sought to examine the extent to which the designs can tolerate mutations (such as those that must be introduced to create a binding surface). Due to the computational expense of the mutational analysis, we focused on the NC_cHLHR_D1 design, mutating each position in sequence to each of alanine, arginine, aspartate, phenylalanine and carrying out a full structure prediction simulation for each. These mutations covered each class of mutation (elimination of the sidechain, introduction of a positive or negative charge, introduction of a bulky aromatic sidechain, or introduction of a small aliphatic sidechain). Mutations preserved chirality (i.e. only D-amino acid to D-amino acid or L-amino acid to L-amino acid mutations were considered). Simulation runs were carried out on the Argonne Leadership Computing Facility’s Blue Gene/Q supercomputer (“Mira”) using a version of the Rosetta simple_cycpep_predict application parallelized using the Message Passing Interface (MPI). The 127 prediction runs (each for a different mutation) required approximately 20,000 CPU-hours apiece, and each produced on the order of 25,000 sampled, closed conformations with good disulfide geometry. For each mutation considered, 50 trajectories were also carried out in which the mainchain was perturbed slightly and relaxed. The resulting collection of samples (from structure prediction and relaxation) was then used to calculate a goodness-of-energy-funnel metric, termed Pnear, by the following expression:
| (1) |
The value of Pnear ranges from 0 (a poor funnel with low-energy alternative conformations or poor sampling close to the design conformation) to 1 (a funnel with a unique low-energy conformation very close to the design conformation). N is the number of samples, and Ei and RMSDi represent the Rosetta score and RMSD from the design structure of the ith sample, respectively. The parameter λ controls how close a state must be to the design if it is to be considered native-like. This was set to 1 Å. Similarly, the parameter kBT governs the extent to which the shallowness or depth of the folding funnel affects the score. This was assigned a value of 1 Rosetta energy unit. The Pnear metric provided a basis for comparison for the mutations considered.
Code availability
All the methods described in this report were implemented in the Rosetta software suite (www.rosettacommons.org). Rosetta software is available free to academic and non-commercial users. Commercial licenses for the suite are available via the University of Washington Technology Transfer Office. Design protocols were implemented using the RosettaScripts interface available within the Rosetta software suite. Input files and command line arguments for each step in our peptide design pipeline are available in the Supplementary Information.
Protein purification of genetically-encodable disulfide-rich peptides
Genes of designed disulfide-rich peptides were cloned into the vector pCDB180 (which we have made available via Addgene) using Gibson Assembly41. Protein expression from E. coli was carried out using a large N-terminal fusion domain consisting of: the native E. coli protein OsmY to direct periplasmic and extracellular localization42, a deca-histidine tag for protein purification, and the SUMO protein Smt3 from Saccharomyces cerevisiae to chaperone folding and provide a mechanism for scarless cleavage of the fusion from the designed protein43. Designed proteins were expressed from BL21*(DE3) E. coli (Invitrogen), and expression cultures were grown overnight with incubation at 37 °C and shaking at 225 RPM. Following expression via Studier autoinduction44, a periplasmic extract45 was prepared by washing cells with: 20% sucrose, 30 mM Tris-HCl pH 8.0, 1 mM ethylenediaminetetraacetic acid pH 8.0, 1 mg/mL lysozyme. Protein was purified from the bacterial-conditioned medium and/or the periplasmic extract by immobilized metal-affinity chromatography (IMAC). During screening, fusion protein was purified from the bacterial-conditioned medium of 50 mL cultures, which typically yielded 9 ± 4 mg of protein (prior to removal of the fusion protein). Protein expression from mammalian cells was carried out using the Daedalus12 system, as previously described in detail. With both purification systems, purified fusion proteins were cleaved by a site-specific proteases, SUMO protease for E. coli and TEV protease for Daedalus, followed by a secondary IMAC step. The final designs were purified to homogeneity by reverse-phase high-performance liquid chromatography on an Agilent 1260 HPLC equipped with a C-18 Zorbax SB-C18 4.6 × 150mm column. Solvent A (Water + 0.1%TFA) and solvent B (Acetonitrile + 0.1%TFA) were run using the following gradient: 0–5% solvent B (5 minutes), 5–45% solvent B(40 minutes).
Synthesis and purification of noncanonical peptides
Linear and cyclic peptides were synthesized as previously described46. Briefly, peptides were synthesized using automated solid phase peptide synthesis with Fmoc (9-fluorenylmethyloxycarbonyl) strategy. Cyclic reduced peptides were obtained after cleavage of the sidechain-protected peptides from the resin, ligation of both termini and the cleavage of sidechain protecting groups. Linear reduced peptides were collected by cleaving the sidechain protecting groups and resin from the peptides simultaneously. All linear or cyclic reduced peptides were oxidized at room temperature in a buffer containing 0.1 M NH4HCO3, where the peptide concentration was 0.25 mg/mL. After 48 h, the mixture was acidified with trifluoroacetic acid, loaded onto a semi-preparative column and purified by RP-HPLC.
Mass spectrometry
Intact samples for each genetically-encodable peptide were diluted in loading buffer with 0.1% formic acid and analyzed on a Thermo Scientific Orbitrap Fusion Tribrid Mass Spectrometer via data-dependent acquisition. Liquid chromatography consisted of a 60 minute gradient across a 15 cm column (75 μm internal diameter) packed with C18 resin with a 3 cm kasil frit trap (150 μm internal diameter) packed with C12 resin. For disulfide connectivity analysis, peptides were digested with sequencing grade modified trypsin (Promega) at 1:50, enzyme to substrate, concentration for 1 hour at 37°C then desalted via mixed-mode cationic exchange (MCX). Peptide samples were dried under vacuum and resuspended in 0.1% formic acid. Digested samples were analyzed using both data-dependent acquisition and targeted methods.
Thermal and chemical denaturation experiments
Circular dichroism (CD) wavelength and temperature scans were recorded on AVIV model 420 or Jasco J-1500 CD spectrometer. For thermal denaturation, peptides samples were prepared at 0.07–0.2 mg/ml final concentration in 10 mM sodium phosphate buffer (pH 7.0). Wavelength scans from 195 nm to 260 nm were recorded at 25 °C, 55 °C, 95 °C, and again after cooling back to 25 °C. For chemical denaturation experiments, samples for each peptide were prepared in the presence of 0 M to 6 M GdnHCl concentrations. The concentration of GdnHCl was measured by refractometry47. Peptide samples were also prepared in the presence of 2.5 mM TCEP (TCEP was pre-equilibrated to pH 7.0 prior to addition), and incubated for 3 hours. Peptide concentrations were the same across all samples. Wavelength scans from 190 nm to 260 nm were recorded for each sample in 0.1 cm cuvette.
NMR analysis and structure determination of genetically-encodable disulfide-rich peptides
Agilent NMR spectrometers operating at 1H resonance frequencies between 500 to 750 MHz equipped with 1H{15N,13C} probes were used to acquire NMR data for gEHE_06, gEEHE_02, gEEH_04, and gHHH_06. The peptides were all uniformly 15N-labeled with gEEH_04 and gHHH_06 also ~10% labeled with 13C. The peptides were suspended in 50 mM sodium chloride, 20 mM sodium acetate, pH 4.8 (gEHE_06 and gEEHE_02) or 50 mM sodium phosphate, 4 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid, 0.02% sodium azide, pH 6.0 (gEEH_04 and gHHH_06) at concentrations between 1.5 and 0.5 mM. The 1H, 13C, and 15N chemical shifts of the backbone and sidechain resonances were assigned by analysis of two-dimensional [15N,1H] HSQC, [13C,1H] HSQC (aliphatic and aromatic), [1H,1H] TOCSY, and [1H,1H] NOESY spectra, and three-dimensional (3D) 15N-resolved [1H,1H] TOCSY, 15N-resolved [1H,1H] NOESY, HNCA, HNCO, and HNHA spectra acquired at 20 °C (for gEHE_06 and gEEHE_02) and 25 °C (gEEH_04 and gHHH_06), respectively. Mixing times of 90 ms (gEHE_06 and gEEHE_02) and 200 ms (gEEH_04 and gHHH_06) were used for 2D and 3D NOESY, respectively. Slowly exchanging amides were identified for gEHE_06 and gEEHE_02 by lyophilizing a 15N-labeled protein, re-dissolving in D2O, and collecting a 2D [15N,1H] HSQC spectrum ~10 minutes after re-dissolving the protein. The resulting D2O sample was subsequently used to collect additional 2D [1H-1H] TOCSY and [1H-1H] NOESY data. Stereospecific assignments for the Val and Leu methyl groups were obtained for gEEH_04 for the 10% fractionally 13C-labelled sample48,49. Because it was not economical to prepare uniformly 13C-labelled peptides by autoinduction, established triple-resonance NMR backbone assignment protocols could not be used. Instead, the carbon resonances were assigned by analyzing the 2D [1H,1H] TOCSY spectra along with [13C,1H] HSQC spectra (collected at natural 13C abundance for gHHH_06, gEHE_06 and gEEHE_02). For gEEH_04, which was 10% fractional 13C-labeled, the assignments were complemented with HNCA spectra. NMR data were processed using the Felix2007 (MSI, San Diego, CA) and PROSA (v6.4) programs and were analyzed using the programs Sparky (v3.115), XEASY, or CARA. Proton chemical shifts were referenced to internal DSS, while 13C and 15N chemical shifts were referenced indirectly via gyromagnetic ratios. Chemical shifts, NOESY peak lists and time domain NMR data were deposited in the BioMagResBank (for accession numbers see Supplementary Table 2-1).
Isotropic overall rotational correlation times of 1.6 - 1.3 ns were inferred from averaged backbone 15N spin relaxation times (www.nmr2.buffalo.edu/nesg.wiki), indicating that all peptides are monomeric in solution. The 1H, 13C, and 15N chemical shift assignments and NOESY peak lists were used for iterative structure calculations using the program CYANA (v 2.1 and 3.97). Chemical shifts were used to derive dihedral phi and psi angle constraints using the program TALOS+50 for residues located in well-defined regular secondary structure elements. For the final structure calculation, hydrogen bond restraints13 were also introduced for gEHE_06 and gEEHE_02, for slowly exchanging amide protons. The resulting ensemble of 20 CYANA conformers was refined by restrained molecular dynamics in an ‘explicit water bath’ using the program CNS (v1.3)51. Structural quality was assessed using the online Protein Structure Validation Suite (PSVS, v1.5)52. The structural statistics are summarized in Supplementary Table 2-1. The coordinates for the 20 conformers representing the solution structures were deposited in the PDB (for accession numbers see Supplementary Table 2-1).
NMR analysis and structure determination of noncanonical peptides
Each noncanonical peptide (1 mg) was dissolved in 500 mL of 10% D2O/90% H2O or 100% D2O (~pH 4). NMR spectra were recorded at 298K on a Bruker Avance-600 spectrometer. Two-dimensional NMR experiments included TOCSY with an 80 s MLEV-17 spin lock, NOESY (200 ms mixing time), ECOSY, as well as natural-abundance 13C and 15N HSQC. Solvent suppression was achieved using excitation sculpting. Spectra were processed using Topspin 2.1 then analysed using CcpNmr Analysis53. Chemical shifts were referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate (DSS).
Initial structures were generated using CYANA and were based upon distance restraints derived from NOESY spectra recorded in both 10% and 100% D2O. The following restraints were also included: disulfide bonds, hydrogen bonds as indicated by slow D2O exchange and sensitivity of amide proton chemical shift to temperature, chi1 restraints from ECOSY and NOESY data, and backbone phi and psi dihedral angles generated using the program TALOS-N54. The final set of structures was generated within CNS55 using torsion angle dynamics, refinement and energy minimization in explicit solvent and protocols as developed for the RECOORD database56. Final structures were assessed for stereochemical quality using MolProbity57.
X-ray crystallography
The gEHEE_06 peptide was purified by size exclusion chromatography on an AKTA Pure using a GE HiLoad 16/600 Superdex 75 pg column, concentrated to 50 mg/ml, and crystallized by vapor diffusion over well solutions of 100 mM citrate (pH 3.5), and 25% PEG3350. Selected crystals were transferred to a cryo-solution of 100 mM citrate (pH 3.5), 20% PEG3350, and 15% glycerol. Diffraction data were collected on a Rigaku Micromax-007HF with a Saturn944+ CCD detector, and integrated and scaled with HKL-2000. Initial phases were determined by molecular replacement using Phaser58 as implemented in the CCP4 software suite with coordinates derived from a Rosetta model for the scaffold. Molecular replacement found 2 molecules per asymmetric unit (ASU). This solution was iteratively refined with the program Refmac followed by model building with COOT, yielding a crystallographic R-values (Rcryst = 39.9%, Rfree = 42.5%). Based on the Matthews’ coefficient, the crystals should have contained 3 molecules per ASU in order to have a reasonable solvent content of 45%. At this point positive electron density appeared that allowed for the manual positioning of a third molecule in the ASU and improving the R-values (Rcryst = 32.0%, Rfree = 34.9%). The model was further improved by including solvent molecules and TLS refinement. The quality of the final model was assessed using ProCheck and Molprobity (overall score: 100th percentile). The final model has been deposited in the PDB with accession code 5JG9. Crystallographic statistics are reported in Supplementary Table S2-2.
Surface redesign
In attempt to reduce solubility and enhance crystallization, we redesigned solvent-exposed residues of designs representing each major topological category (mixed α/β, all β-sheet, all α-helical). Two resurfaced variants were selected for each design bearing between one to two solvent-exposed tyrosine residues. We then expressed and purified these resurfaced designs using Daedalus, all of which expressed solubly and exhibited a redox-sensitive migration time by reverse-phase HPLC. We were only able to obtain diffracting protein crystals for redesign gEEHE_2.1_02_0008, which diffracted to 2.90 Å resolution (Supplementary Table 2-2). However, Matthews calculations predicted non-crystallographic symmetry with approximately nineteen copies in the asymmetric unit, and attempts to phase the crystal by molecular replacement were unsuccessful, as were attempts at reproducing the crystal outside of the initial screen.
Extended Data
Extended Data Figure 1. Disulfide bonds are well defined by X-ray crystallography.

An Fo – Fc omit-map is shown contoured at 4 σ for design gEHEE_06. Disulfide sulfur atoms were removed, and the omit-map was calculated following real-space refinement.
Extended Data Figure 2. Flowchart of pipelines for designing noncanonical cyclic peptides.

Inputs are shown in blue, RosettaScripts-automated parts of the pipeline are in green, parts carried out by Rosetta standalone applications are pink (the fragment picker application) and purple (the various structure prediction applications), parts performed with molecular dynamics software are yellow, and manual steps are grey. a) Fragment assembly-based design pipeline. Final computational validation was carried out using MD simulations and fragment-based Rosetta ab initio structure prediction. For peptides containing isolated D-amino acids, these residues were mutated to glycine for Rosetta ab initio structure prediction. b) Fragment-free, GenKIC-based design pipeline. This approach permits design of noncanonical topologies like the mixed HLHR topology, which occurs in no known natural protein. The GenKIC-based structure prediction algorithm is described in Extended Data Figure 7 and in the Supplementary Information.
Extended Data Figure 3. Sidechain placement in noncanonical peptide designs chosen for experimental characterization.

Designs are shown as cartoon and stick representations (top row in each box) and as van der Waals spheres showing sidechain packing (bottom row in each box). L-amino acid residues are shown in cyan, and D-amino acid residues are coloured orange. Sidechains of D- or L-variants of alanine, phenylalanine, isoleucine, leucine, valine, tryptophan, and tyrosine are coloured grey to aid visualization of hydrophobic packing interactions.
Extended Data Figure 4. Molecular dynamics screening of designed peptides.

Fifty independent molecular dynamics (MD) simulations in explicit solvent conditions, all starting from the designed peptide, were used for discriminating good, kinetically-stable (e.g. EHE_D1) designs from non-optimal designs of the same topology (e.g. EHE_X18 and EHE_X11). a) Five representative trajectories from MD simulation runs. Designs that showed good convergence, and smaller fluctuations were selected for further experimental characterization. b) RMSD distribution from all 50 trajectories. Only the last one-third of the trajectory was used for this analysis. Designs with narrower distributions were picked for further testing. c) Concatenated trajectory of all 50 independent runs show lower fluctuations for the more optimal designs.
Extended Data Figure 5. Structural characterization of NC_EEH_D1.

NMR structure of NC_EEH_D1 does not match the designed topology. a) Rosetta-designed model for NC_EEH_D1. b) Ensemble of conformers representing the NMR solution structure. c) Superposition of the designed model (blue) with a representative NMR conformer (green).
Extended Data Figure 6. Structural mapping of sequence-aligned region between NC_EHE_D1 and 2MA5.

Design NC_EHE_D1 and PDB entry 2MA5 show weak but significant (e-value: 2×10−4) sequence alignment, which is highlighted in purple. The aligned region folds into very different structures in the different contexts of peptide and protein.
Extended Data Figure 7. Generalized kinematic closure (GenKIC) algorithm flowchart.

GenKIC allows sampling of closed conformations of arbitrary chains of atoms, passing through canonical or noncanonical backbone or sidechain linkages. Bond length, bond angle, and torsional degrees of freedom in the chain can be fixed, perturbed from a starting value by small amounts, set to user-defined values, or sampled randomly. The algorithm then solves for six torsion angles adjacent to three user-defined pivot atoms in order to enforce closure of the loop. The many solutions from the closure are then filtered internally, and each can be subjected to arbitrary user-defined Rosetta protocols and filtration in order to prune the solution list further. A single solution is selected from those passing filters by a user-defined selection criterion. This flowchart shows the steps in a single invocation of the algorithm; for sampling, a user may specify that the algorithm be applied any number of times. User inputs are shown in blue, steps carried out by the GenKIC algorithm itself are in green, steps carried out by Rosetta code external to the GenKIC algorithm are shown in yellow, and outputs are shown in salmon.
Extended Data Figure 8. A new fragment-free structure prediction algorithm.

a) Flowchart diagramming the steps to generate a single sampled conformation. In typical usage, this process would be repeated tens of thousands of times to produce many samples. Inputs (the peptide sequence and an optional PDB file for the design structure) are shown in blue, and outputs (the sampled structure, its energy, and its RMSD from the design structure) are shown in salmon. Steps performed by the Generalized Kinematic Closure algorithm are shaded green, and setup and completion steps performed by the simple_cycpep_predict application are shown in yellow. Further details of this algorithm are discussed in the Supplementary Information available online. b) The initial, random peptide conformation with bad terminal peptide bond geometry. c) Ensemble of closed conformations found for a single closure attempt. In this example, residue 7 (cyan) is the fixed anchor residue. Certain regions of the peptide have been set to left- or right-handed helical conformations prior to solving closure equations. d) A single closed solution with relative cysteine sidechain orientations that pass the initial, low-stringency filter for disulfide (fa_dslf) conformational energy. e) The resulting structure, following sidechain repacking, energy-minimization, and cyclic de-permutation.
Extended Data Figure 9. Mutational tolerance of selected genetically-encodable designs.

RP-HPLC traces for the parental designs are shown next to the redesigned variants where applicable. Proteins run under oxidized conditions are shown in black while proteins run following reduction with 10mM DTT are shown in red. Insets within each panel are shown only to highlight the SDS-PAGE mobility of each purified protein under oxidizing (left band) and reducing conditions (right band). Sequence alignments are shown with the mutated positions highlight in red, along with theoretical isoelectric points as calculated by ProtParam.
Extended Data Figure 10. Mutational tolerance of selected NC designs.

a–b) Mutational tolerance of D-proline, L-proline loop of design NC_cEE_D1 (green in panel a), assessed by secondary 1Hα chemical shift for the design sequence (black bars in panel b) and the p18d loop mutation (red bars). Eliminating this key proline residue does not result in loss of β-strand signal. c–d) Mutational tolerance of loop region of design NC_HEE_D1 (green in panel c), as assessed by CD spectroscopy for the design sequence (left plot, panel d) and for the D19T, p20q, P21D triple mutant (right plot, panel d). Both proline residues may be mutated without loss of secondary structure or major change in the thermal stability. e–g) Computationally predicted mutational tolerance of design NC_HLHR_D1, across the entire sequence. Each position was successively mutated in silico to D- or L-alanine, arginine, aspartate, phenylalanine, or valine (preserving the position’s chirality), and full folding simulations were carried out with the Rosetta simple_cycpep_predict application. Folding funnel quality was evaluated using the Pnear metric described in the Methods. e) Representative plots of energy vs. RMSD from the design structure, plotted for the design sequence (top), for the non-disruptive R14F mutation (middle), and for the e18v mutation (bottom). Results from GenKIC-based structure prediction runs are shown in blue, and relaxation runs, in orange. Note that the bottom case shows many sampled states far from the design state with energy equal to or less than the design state energy. f) Mutational tolerance by position (vertical axis) and mutation (horizontal axis). Blue rectangles represent well-tolerated mutations, and red to black rectangles represent disruptive mutations, based on Pnear evaluation of the folding funnel. Black borders indicate the design sequence. g) Mutational tolerance mapped onto the NC_HLHR_D1 structure, with colours as in the previous panel. Most positions tolerate mutation well, with only the disulfide bridge (C8-c21) and the salt bridges formed by e18 being highly sensitive. The hydrogen bond networks formed by residues Q5, e24, and s25 show some moderate sensitivity to mutation, as do residues E3 and e16.
Supplementary Material
Acknowledgments
Computer time was awarded by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program. This research used resources of the Argonne Leadership Computing Facility, a Department of Energy (DOE) Office of Science User Facility supported under Contract DE-AC02-06CH11357. We thank the University of Washington Hyak supercomputing network for computing and data storage resources, and Rosetta@Home volunteer participants on BOINC for additional computing resources. We are grateful for facility access at the Queensland NMR Network. We thank Darwin Alonso, James Bardwell, Gira Bhabha, TJ Brunette, Damian Ekiert, Alexander Ford, Nicholas Hasle, Brandon Keir, Nobu Koga, Yuan Liu, Dean Madden, Binchen Mao, Damon May, Victor Ovchinnikov, Sanjay Srivatsan, Lance Stewart, Ruud van Deursen, and Miriam Williamson for help and advice, and Ratika Krishnamurty, Parisa Hosseinzadeh, and Anastassia Vorobieva for critical comments and manuscript suggestions. This work was supported by NIH grant P50 AG005136 supporting the Alzheimer’s Disease Research Center, philanthropic gifts from the Three Dreamers and Washington Research Foundation, and funding from the Howard Hughes Medical Institute. The Australian Research Council funds DJC as an Australian Laureate Fellow (FL150100146). CDB was supported by NIH grant T32-H600035. TS acknowledges NIH support (GM094597), and SVSRKP, AE and XX were supported with NESG funds. EC is funded by NIGMS GM090205. We thank Peter Rupert and Roland K. Strong at the Fred Hutchinson Cancer Research Center for aid collecting and refining X-ray data for gEHEE_06. GWB was funded by the National Institute of Allergy and Infectious Diseases, National Institute of Health, Department of Health and Human Services (Federal Contract HHSN272201200025C). Some of this research was performed at the W.R. Wiley Environmental Molecular Sciences Laboratory, located at Pacific Northwest National Laboratory (PNNL) and sponsored by the DOE’s Office of Biological and Environmental Research program. Battelle operates PNNL for the DOE.
Footnotes
Author Contribution
CDB, GB, VKM and DB designed the study. VKM developed algorithms with help from AW, EC, YS, GB, RB, CDB, GJR, and TWL. CDB and JMG designed canonical peptides with help from DB, GJR, and TWL. GB designed heterochiral and backbone-cyclized peptides with help from VKM, DB, PG, and PSH. CDB expressed and characterized designed canonical peptides from E. coli with help from JMG and SAR. JMG performed MS analysis. WAG and CEC purified canonical peptides via Daedalus and determined X-ray crystal structures. GWB, SVSRKP, AE, and TS determined NMR solution structures of canonical peptides, purified with isotopic labeling by CDB. OC and GB synthesized, purified and characterized designed noncanonical peptides. PJH and DJC determined NMR solution structures of noncanonical peptides. PJH, QK and DJC analysed data from structure determination of noncanonical peptides. CDB, GB, VKM, and DB wrote the manuscript with help from all authors.
NMR solution structures are deposited to RCSB Protein Data Bank with accession codes 5JG9, 2ND2, 2ND3, 3JHI, 5JI4, 5KVN, 5KWO, 5KWP, 5KWX, 5KX2, 5KWZ, 5KW1, 5KX0.
Authors declare no competing financial interests.
References
- 1.Conibear AC, et al. Approaches to the stabilization of bioactive epitopes by grafting and peptide cyclization. Biopolymers. 2016;106:89–100. doi: 10.1002/bip.22767. [DOI] [PubMed] [Google Scholar]
- 2.Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 3.Góngora-Benítez M, Tulla-Puche J, Albericio F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem Rev. 2014;114:901–926. doi: 10.1021/cr400031z. [DOI] [PubMed] [Google Scholar]
- 4.Kimura RH, Levin AM, Cochran FV, Cochran JR. Engineered cystine knot peptides that bind alphavbeta3, alphavbeta5, and alpha5beta1 integrins with low-nanomolar affinity. Proteins. 2009;77:359–369. doi: 10.1002/prot.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyken SE, et al. De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity. Science. 2016;352:680–687. doi: 10.1126/science.aad8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunette TJ, et al. Exploring the repeat protein universe through computational protein design. Nature. 2015;528:580–584. doi: 10.1038/nature16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin YR, et al. Control over overall shape and size in de novo designed proteins. Proc Natl Acad Sci U S A. 2015;112:E5478–85. doi: 10.1073/pnas.1509508112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doyle L, et al. Rational design of α-helical tandem repeat proteins with closed architectures. Nature. 2015;528:585–588. doi: 10.1038/nature16191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koga N, et al. Principles for designing ideal protein structures. Nature. 2012;491:222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leaver-Fay A, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang PS, et al. High thermodynamic stability of parametrically designed helical bundles. Science. 2014;346:481–485. doi: 10.1126/science.1257481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bandaranayake AD, et al. Daedalus: a robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res. 2011;39:e143. doi: 10.1093/nar/gkr706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagaram US, et al. Structural and functional studies of a phosphatidic acid-binding antifungal plant defensin MtDef4: identification of an RGFRRR motif governing fungal cell entry. PLoS One. 2013;8:e82485. doi: 10.1371/journal.pone.0082485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G, et al. NMR data collection and analysis protocol for high-throughput protein structure determination. Proc Natl Acad Sci U S A. 2005;102:10487–10492. doi: 10.1073/pnas.0504338102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma D, Rajarathnam K. 13C NMR chemical shifts can predict disulfide bond formation. J Biomol NMR. 2000;18:165–171. doi: 10.1023/a:1008398416292. [DOI] [PubMed] [Google Scholar]
- 16.Richardson JS. β-Sheet topology and the relatedness of proteins. Nature. 1977;268:495–500. doi: 10.1038/268495a0. [DOI] [PubMed] [Google Scholar]
- 17.Syud FA, Stanger HE, Gellman SH. Interstrand side chain--side chain interactions in a designed beta-hairpin: significance of both lateral and diagonal pairings. J Am Chem Soc. 2001;123:8667–8677. doi: 10.1021/ja0109803. [DOI] [PubMed] [Google Scholar]
- 18.Lai JR, Huck BR, Weisblum B, Gellman SH. Design of non-cysteine-containing antimicrobial beta-hairpins: structure-activity relationship studies with linear protegrin-1 analogues. Biochemistry. 2002;41:12835–12842. doi: 10.1021/bi026127d. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Yadav V, Smart AL, Tajiri S, Basit AW. Toward oral delivery of biopharmaceuticals: an assessment of the gastrointestinal stability of 17 peptide drugs. Mol Pharm. 2015;12:966–973. doi: 10.1021/mp500809f. [DOI] [PubMed] [Google Scholar]
- 20.Coutsias EA, Seok C, Jacobson MP, Dill KA. A kinematic view of loop closure. J Comput Chem. 2004;25:510–528. doi: 10.1002/jcc.10416. [DOI] [PubMed] [Google Scholar]
- 21.Mandell DJ, Coutsias EA, Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat Methods. 2009;6:551–552. doi: 10.1038/nmeth0809-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trabi M, Schirra HJ, Craik DJ. Three-dimensional structure of RTD-1, a cyclic antimicrobial defensin from Rhesus macaque leukocytes. Biochemistry. 2001;40:4211–4221. doi: 10.1021/bi002028t. [DOI] [PubMed] [Google Scholar]
- 23.Sia SK, Kim PS. A designed protein with packing between left-handed and right-handed helices. Biochemistry. 2001;40:8981–8989. doi: 10.1021/bi010725v. [DOI] [PubMed] [Google Scholar]
- 24.Renfrew PD, Douglas Renfrew P, Choi EJ, Richard B, Brian K. Incorporation of Noncanonical Amino Acids into Rosetta and Use in Computational Protein-Peptide Interface Design. PLoS One. 2012;7:e32637. doi: 10.1371/journal.pone.0032637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drew K, et al. Adding diverse noncanonical backbones to rosetta: enabling peptidomimetic design. PLoS One. 2013;8:e67051. doi: 10.1371/journal.pone.0067051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleishman SJ, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang PS, et al. RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One. 2011;6:e24109. doi: 10.1371/journal.pone.0024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J, Lee D, Park H, Coutsias EA, Seok C. Protein loop modeling by using fragment assembly and analytical loop closure. Proteins. 2010;78:3428–3436. doi: 10.1002/prot.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrison PM, Sternberg MJ. Analysis and classification of disulphide connectivity in proteins. The entropic effect of cross-linkage. J Mol Biol. 1994;244:448–463. doi: 10.1006/jmbi.1994.1742. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Granillo A, Annavarapu S, Zhang L, Koder RL, Nanda V. Computational design of thermostabilizing D-amino acid substitutions. J Am Chem Soc. 2011;133:18750–18759. doi: 10.1021/ja205609c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Meara MJ, et al. Combined Covalent-Electrostatic Model of Hydrogen Bonding Improves Structure Prediction with Rosetta. J Chem Theory Comput. 2015;11:609–622. doi: 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradley P, Misura KMS, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 33.Caves LS, Evanseck JD, Karplus M. Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci. 1998;7:649–666. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wijma HJ, et al. Computationally designed libraries for rapid enzyme stabilization. Protein Eng Des Sel. 2014;27:49–58. doi: 10.1093/protein/gzt061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Götz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 12. University of California; San Francisco: 2012. [Google Scholar]
- 36.Jorgensen WL, Corky J. Temperature dependence of TIP3P, SPC, and TIP4P water from NPT Monte Carlo simulations: Seeking temperatures of maximum density. J Comput Chem. 1998;19:1179–1186. [Google Scholar]
- 37.Loncharich RJ, Brooks BR, Pastor RW. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers. 1992;32:523–535. doi: 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- 38.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089. [Google Scholar]
- 39.Ryckaert JP, Giovanni C, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 40.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 41.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 42.Kotzsch A, et al. A secretory system for bacterial production of high-profile protein targets. Protein Sci. 2011;20:597–609. doi: 10.1002/pro.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marblestone JG, et al. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 2006;15:182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 45.Neu HC, Heppel LA. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J Biol Chem. 1965;240:3685–3692. [PubMed] [Google Scholar]
- 46.Cheneval O, et al. Fmoc-based synthesis of disulfide-rich cyclic peptides. J Org Chem. 2014;79:5538–5544. doi: 10.1021/jo500699m. [DOI] [PubMed] [Google Scholar]
- 47.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 48.Neri D, et al. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional carbon-13 labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 49.Herve du Penhoat C, et al. [Accessed: 13th July 2016];The NMR solution structure of the 30S ribosomal protein S27e encoded in gene RS27_ARCFU of Archaeoglobus fulgidis reveals a novel protein fold. doi: 10.1110/ps.03589204. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15096641. [DOI] [PMC free article] [PubMed]
- 50.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Linge JP, Williams MA, Spronk CAEM, Alexandre MJ, Michael N. Refinement of protein structures in explicit solvent. Proteins: Struct Funct Bioinf. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 52.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66:778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 53.Vranken WF, et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins: Struct Funct Bioinf. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 54.Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR. 2013;56:227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 56.Nederveen AJ, et al. RECOORD: A recalculated coordinate database of 500 proteins from the PDB using restraints from the BioMagResBank. Proteins: Struct Funct Bioinf. 2005;59:662–672. doi: 10.1002/prot.20408. [DOI] [PubMed] [Google Scholar]
- 57.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.