

Abstract
Background and Aims
To date chloroplast genomes are available only for members of the non-protein amino acid-accumulating clade (NPAAA) Papilionoid lineages in the legume family (i.e. Millettioids, Robinoids and the ‘inverted repeat-lacking clade’, IRLC). It is thus very important to sequence plastomes from other lineages in order to better understand the unusual evolution observed in this model flowering plant family. To this end, the plastome of a lupine species, Lupinus luteus, was sequenced to represent the Genistoid lineage, a noteworthy but poorly studied legume group.
Methods
The plastome of L. luteus was reconstructed using Roche-454 and Illumina next-generation sequencing. Its structure, repetitive sequences, gene content and sequence divergence were compared with those of other Fabaceae plastomes. PCR screening and sequencing were performed in other allied legumes in order to determine the origin of a large inversion identified in L. luteus.
Key Results
The first sequenced Genistoid plastome (L. luteus: 155 894 bp) resulted in the discovery of a 36-kb inversion, embedded within the already known 50-kb inversion in the large single-copy (LSC) region of the Papilionoideae. This inversion occurs at the base or soon after the Genistoid emergence, and most probably resulted from a flip–flop recombination between identical 29-bp inverted repeats within two trnS genes. Comparative analyses of the chloroplast gene content of L. luteus vs. Fabaceae and extra-Fabales plastomes revealed the loss of the plastid rpl22 gene, and its functional relocation to the nucleus was verified using lupine transcriptomic data. An investigation into the evolutionary rate of coding and non-coding sequences among legume plastomes resulted in the identification of remarkably variable regions.
Conclusions
This study resulted in the discovery of a novel, major 36-kb inversion, specific to the Genistoids. Chloroplast mutational hotspots were also identified, which contain novel and potentially informative regions for molecular evolutionary studies at various taxonomic levels in the legumes. Taken together, the results provide new insights into the evolutionary landscape of the legume plastome.
Keywords: Lupinus luteus, European yellow lupine, legume, Genistoid clade, chloroplast genome evolution, structural plastid rearrangement, 36-kb inversion, inverted repeats, flip–flop recombination, lineage-specific marker, functional gene transfer, Papilionoidae, repeated plastid sequences, sequence divergence, plastome hotspots, Fabaceae phylogeny
INTRODUCTION
Legumes (Fabaceae) are the third largest angiosperm family, with 727 genera and about 20 000 species (Lewis et al., 2005). They are characterized by a wide biological and ecological diversity (Cronk et al., 2006), and they are of great economic importance, particularly for human consumption or as animal forage. This family is composed of two main groups (Fig. 1): Caesalpinioideae sensu lato (s.l.; including Mimosoideae) and Papilionoideae (Wojciechowski et al., 2004; Cardoso et al., 2012). The Papilionoideae is divided into six major clades: the Genistoids, Dalbergioids, Mirbelioids, Millettioids, Robinioids and the inverted repeat lacking clade (IRLC) (Cronk et al., 2006). Within the Genistoids, Lupinus displays particular functional properties compared with other legumes, such as active nitrogen metabolism and the production of allelopathic substances of ecological and agronomical interest (Guillon and Champ, 2002; Magni et al., 2004; Pilvi et al., 2006). Additionally, Lupinus is the only known legume that does not form mycorrhizal symbioses (Sprent, 2007). They also have considerable potential for phytoremediation due to their ability to metabolize nitrogen pollutants such as atrazine (Garcinuno et al., 2003).
Fig. 1.

Simplified phylogenetic tree representing the main clades circumscribed in the legume family [redrawn from Wojciechowski et al. (2004) and Cardoso et al. (2012)]. Filled triangles indicate lineages for which one or more whole chloroplast genome sequences are available, whereas open triangles indicate lineages for which no plastome sequence exists, including the core Genistoid clade (boxed) targeted in this study.
Plastome organization is highly conserved among most flowering plants (Jansen and Ruhlman, 2012), most having a quadripartite structure composed of two copies of an inverted repeat (IR) separated by large and small single-copy regions (LSC and SSC). However, a few angiosperm families, including the Fabaceae, present an unusual plastome structure and evolution. In this family, the loss of one IR in the Papilionoideae (Wojciechowski et al., 2004), the presence of many repetitive sequences (Saski et al., 2005; Magee et al., 2010), the occurrence of relatively large inversions (Palmer and Herbon, 1988; Perry et al., 2002; Magee et al., 2010) and the presence of a localized hypermutable region (Magee et al., 2010) have been detected. Aberrant DNA repair was inferred as a possible cause for these plastomic rearrangements and accelerated rates of nucleotide substitutions (Jansen et al., 2007).
Although most photosynthetic angiosperm plastomes contain 79 protein-coding genes, various derived lineages exhibit slightly fewer genes. Most of these rare chloroplastic gene losses occurred in species whose plastomes are highly rearranged relative to the ancestral angiosperm plastome (Jansen et al., 2007). Since the emergence of the Fabaceae, there has been loss of five different chloroplastic genes: accD, psaI, rpl23, rps16 and ycf4 (Jansen et al., 2007; Magee et al., 2010). It is very likely that the genes lost from the plastome were previously functionally transferred to the nucleus or replaced by a nuclear gene of prokaryotic or eukaryotic origin. For example, the chloroplast accD gene was functionally transferred to the nucleus in Trifolium species (Magee et al., 2010) and the plastidic rps16 gene was functionally replaced by a nuclear-encoded rps16 gene of mitochondrial origin in Medicago truncatula (Ueda et al., 2008).
During the last decade, knowledge of the organization and evolution of legume plastomes has rapidly expanded with the development of next-generation sequencing (NGS) technologies. Ten legume plastomes have now been sequenced: Cicer arietinum (Jansen et al., 2008), Glycine max (Saski et al., 2005), Lathyrus sativus (Magee et al., 2010), Lotus japonicus (Kato et al., 2000), M. truncatula (NC_003119), Millettia pinnata (Kazakoff et al., 2012), Phaseolus vulgaris (Guo et al., 2007), Pisum sativum (Magee et al., 2010), Trifolium subterraneum (Cai et al., 2008) and Vigna radiata (Tangphatsornruang et al., 2010). The sequencing of these plastomes confirmed previous observations of major rearrangements in this family, including a 50-kb inversion present in most papilionoids (Palmer and Thompson 1982; Lavin et al., 1990; Doyle et al., 1996; Wojciechowski et al., 2004; Jansen et al., 2008) and the loss of one copy of the IR region in one of the papilionoid clade, called the IRLC (Palmer and Thompson, 1982; Lavin et al., 1990; Wojciechowski et al., 2004; Jansen et al., 2008). However, all the Papilionoideae plastomes sequenced to date belong to three clades (Millettioids, Robinioids and IRLC) within the non-protein amino acid-accumulating clade (NPAAA clade; according to Cardoso et al., 2012). Thus it is essential to investigate representatives from other Papilionoid lineages to better understand plastome evolution within the Papilionoideae, and more broadly within legumes. In this context, the genus Lupinus is a good candidate to represent the core Genistoids (Wojciechowski et al., 2004; Cronk et al., 2006; Cardoso et al., 2012) that is one of the poorly studied legume lineages. Although considerable strides have been made in elucidating the evolutionary history of the Fabaceae using plastid DNA sequence-based phylogenies (Wojciechowski et al., 2004; Cardoso et al., 2012), there is still a great need to elucidate more accurately relationships at other taxonomic levels among and within lineages of the 50-kb-inversion Papilionoid clade, including within the Genistoids and in the genus Lupinus (Ainouche and Bayer, 1999; Ainouche et al., 2004; Hughes and Eastwood, 2006; Drummond, 2008; Mahé et al., 2011a, b). Therefore, the lupine plastome sequence not only provides the raw material to extend understanding of legume genome organization and evolution, but also provides an important source of phylogenetically informative plastid molecular markers, which have the advantage of being uniparentally (maternally) inherited and generally non-recombinant (Jansen et al., 2007; Moore et al., 2007, 2010).
Here we report the complete sequence of the chloroplast genome of Lupinus luteus, the first sequenced in the core Genistoids. After reconstruction and annotation, this genome has been compared with other Fabaceae and extra-Fabales plastomes, allowing the identification of a noteworthy 36-kb inversion. A PCR and sequencing survey of this inversion across various legume representatives provided evidence that this inversion represents a novel genomic rearrangement, characterizing the core Genistoids. The gene content within the L. luteus plastome has also been compared with that of other Fabaceae and closely related species in order to identify chloroplast genes lost from the L. luteus plastome. We verified that the chloroplast genes missing in the Lupinus plastome were functionally transferred to the nucleus. Finally, we evaluated the sequence divergence between the lupine and other Fabaceae plastomes at different levels (exon, intron and intergenic) in order to better understand the unusual plastome evolution and to suggest potentially useful plastid regions for molecular phylogenetic analyses in Fabaceae.
MATERIALS AND METHODS
DNA extraction, high-throughput sequencing and isolation of chloroplast sequences
Genomic DNA was extracted from fresh leaves of an individual sample (Lab. collection ref. number: M6=EGSM6Llu2) from a natural population of Lupinus luteus collected at Bou Tlelis, Oran in Algeria, North Africa. DNA extraction was performed using a NucleoSpin® Plant II kit (Macherey Nagel) following the manufacturer's instructions. The genomic DNA was subjected to two high-throughput methods of sequencing: one run using pyrosequencing with the GS-FLX (454 Life Science, Roche) platform (OSUR/biogenouest; Université de Rennes-1) that generated 799 732 reads of approx. 400 bases, and one flow cell lane performed with an Illumina HiSeq 2000 platform (BGI, Hong Kong) that yielded 11·46 million 2 × 100-base paired-end reads from a library of approx. 500 base DNA fragments. Reads corresponding to plastome sequences were extracted from the Roche-454 data set using a blast similarity search (e-value 10–6, 90 % identity) against the fully sequenced plastomes of G. max (NC_007942), M. truncatula (NC_003119), L. japonicus (NC_002694), C. arietinum (NC_011163), P. sativum (NC_0147057), T. subterraneum (NC_011828), L. sativus (NC_014063), M. pinnata (NC_016708), V. radiata (NC_013843), P. vulgaris (NC_009259), Populus trichocarpa (NC_009143) and Arabidopsis thaliana (NC_000932). A total of 21 018 reads corresponding to plastid sequences were obtained from the 454 sequencing and 509 962 paired-end reads from Illumina.
Plastome assembly and annotation
De novo assembly was performed from filtered Roche-454 reads using Newbler (v. 2.5.3, 454 Life Science). A total of 45 contigs ranging from 450 to 25 000 bases were obtained and organized using the G. max plastome as a reference. Illumina paired-end reads having at least one mate mapping with Bowtie (Langmead et al., 2009) on the 45 contigs were extracted from the Illumina data set. The draft plastome sequence as well as the junctions between contigs were verified and corrected with the 509 962 paired-end Illumina reads extracted using Mira v. 3.4.0 (Chevreux et al., 1999) and Bowtie (Langmead et al., 2009). The 454 and Illumina data sets allowed a 73× (s.d. 53) and 884× coverage (s.d. 466) of the newly reconstructed L. luteus plastome, respectively.
Plastome annotation was conducted in four steps. (1) Identification of protein-coding sequences by aligning (blastp, e-value threshold 10–5) G. max protein-coding sequences obtained from the ChloroplastDB (Cui et al., 2006) against chloroplastic open reading frames (ORFs) extracted from the Lupinus plastome sequence using the perl script get_orf.pl designed by Paul Stothard (University of Alberta). (2) Identification of rRNA and tRNA sequences by direct alignment of G. max tRNAs and rRNAs against the Lupinus plastome sequence. (3) Verification of the identification of all plastomic genes using DOGMA (Wyman et al., 2004). (4) Verification of the annotation by performing manual alignment using BioLign and multiple contig editor (v. 4.0.6.2). A graphical representation of the chloroplast genome was performed using the CIRCOS software (Krzywinski et al., 2009).
To determine the presence of codon bias, the number of codons ending with A–T or C–G was tallied and a χ2 test was performed for each amino acid. These tests were subjected to a Bonferroni correction for multiple testing performed with the R software package (http://www.r-project.org).
Identification of repeat elements
The number and location of repeated elements (tandem, palindrome, dispersed direct and dispersed inverted repeats) in the L. luteus plastome were determined using REPuter (Kurtz et al., 2001). We used the same parameters as previously used for other Fabaceae species (Saski et al., 2005; Cai et al., 2008; Tangphatsornruang et al., 2010). More precisely, we searched for repeated elements with a minimum size of 30 bp and a Hamming distance of 3 (sequence identity of ≥90 %). One copy of the IR was removed before performing the analysis.
Identification and origin of the 36-kb inversion by PCR screening and sequencing
In order to identify the putative presence of large structural variation (>1 kb) within the L. luteus plastome, breaks of synteny were searched between plastomes of L. luteus, other legumes and two outgroup taxa (Cucumis sativus from the Cucurbitales and Prunus persica from the Rosales) by performing dot plots using the Gepard software (Krumsiek et al., 2007).
To determine the origin of the large inversion observed in L. luteus, its presence/absence was surveyed by PCR in Lupinus and in representatives of various genera more or less closely related to the lupines in the core Genistoids: Argyrolobium uniflorum, Chamaecytisus mollis, Crotalaria saharae, Echinospartum boissieri, Genista florida, Genista tricuspidata, Laburnum anagyroides, Lupinus microcarpus, Retama sphaerocarpa, Sophora japonica, Thermopsis rhombifolia and Ulex minor. Outgroup taxa were also screened for the presence/absence of this inversion, for instance: Cercis siliquastrum that is basal in the legume family; Acacia dealbata that belongs to the Mimosoids; and Cladrastis lutea, a Papilionoid that is sister to the 50-kb-inversion clade. A PCR strategy using primer pairs diagnostic for the presence or absence of the inversion was conducted. The primer pairs were designed in either conserved ycf3 and psbI, or rps4 and ycf3 protein-coding sequences, which are flanking the inversion endpoints, to allow the assessment of the presence or absence of the inversion.
Each PCR amplification was performed in a total volume of 50 μL containing 10 μL of 5× Go taq green buffer (Promega), 5 μL of 2 mm deoxyribonucleotide mix, 4 μL of each primer (5 mm), 0·2 μL of Go Taq polymerase (5 U μL–1) and 20 ng of template DNA. Cycling conditions were 94 °C for 2 min, followed by 35 cycles of 94 °C for 45 s, 55 °C for 30 s and 72 °C for 90 s, and a final extension of 72 °C for 7 min. The primer pairs used to detect the absence or presence of the 36-kb inversion were: rps4-bef-F (5′-CAATCAAATAATAGATAGTAAATGGGTTG-3′) and ycf3-bef-R (5′-GGAATTATTCGTAATAATATATTGGCTAC-3′); and ycf3-inv-F (5′- CGTAATAAGATATTGGCTAC-3′) and psbI-int-R (5′-CTCTTTTCATCTTCGGATTC-3′). The PCR products were then purified using the NucleoSpin Gel and PCR Clean-up purification kit (Macherey-Nagel) and sequenced directly in both directions (Macrogen Europe, Amsterdam).
Evolution of the gene content in the Fabaceae plastome and identification of genes functionally transferred to the nucleus in Lupinus
In order to determine whether L. luteus has recently lost chloroplastic genes, its plastome was compared with those of ten other legume species (M. pinnata, V. radiata, G. max, P. vulgaris, T. subterraneum, M. truncatula, L. japonicas, C. arietinum, P. sativum and L. sativus) and two ougroup species available in GenBank. During Fabaceae evolution, five chloroplastic genes (accD, psaI, rpl22, rpl23 and rps16) have been lost from the plastome of various lineages, of which three (accD, rpl22 and rps16) were shown to have been independently functionally relocated to the nucleus or replaced by a nuclear gene in different Fabaceae (Gantt et al., 1991; Millen et al., 2001; Ueda et al., 2008; Magee et al., 2010). We searched for putative functional transfer to the nucleus (functional relocation or intermediate stage) of these five plastid genes within transcriptomic data available from our laboratory for Lupinus mariae josephi (unpubl. data). The identification of these putative functional transfers was performed by blasting (e-value threshold: 10–10) the following sequences against the transcripts of L. mariae josephi: the Trifolium repens nuclear accD (Magee et al., 2010) and the L. luteus plastidic accD genes; the P. sativum nuclear rpl22 sequence (Gantt et al., 1991); the plastidic psaI, rpl23 and ycf4 genes from various Fabaceae (L. luteus, L. japonicus and P. vulgaris); and the M. truncatula nuclear-encoded rps16 genes of mitochondrial origin (Ueda et al., 2008). The presence of a transit peptide-encoding sequence within the identified chloroplastic genes functionally replaced in the nucleus was then predicted using BaCelLo (Pierleoni et al., 2006), Predotar (Small et al., 2004) and TargetP (Emanuelsson et al., 2000) software programs. To confirm that the nuclear rpl22 gene identified in L. mariae josephi results from an early functional transfer to the nucleus in the common ancestor of all flowering plants, as demonstrated with P. sativum by Gantt et al. (1991), we aligned these sequences (Lupinus and Pisum) with the rpl22 amino acid sequences from eubacteria, algae, bryophytes and land plants using the Geneious package (http://www.geneious.com/). After excluding the extreme 5′ and 3′ ends of the sequences, a data matrix of 98 amino acids was subjected to phylogenetic analyses using PHYML (Guindon and Gascuel, 2003) and Neighbor–Joining (Saitou and Nei, 1987). The tree was rooted using the eubacteria Mycoplasma. Bootstrap values were performed with 1000 replicates (Felsenstein, 1985).
Evaluating sequence divergence between the complete lupine plastid genome and those from other legumes and Fabids
Sequence divergence between L. luteus and ten other Fabaceae plastomes was evaluated independently for each homologous regions aligned with MUSCLE (Edgar, 2004). Pairwise distances were calculated with the ape R-cran Package (Paradis et al., 2011, available at http://cran.r-project.org/web/packages/ape/ape.pdf) using the Kimura-2-parameters (K2p) evolution model (Kimura, 1980). The mean sequence divergence rate of the different genetic categories [i.e. intergenic spacers (IGSs), introns, rRNA and tRNA, and exons] was compared using Mann–Whitney test with Bonferroni correction. Additionally, sequence divergence of coding-protein genes (exons) was estimated using the synonymous (Ks) and non-synonymous (Ka) nucleotide substitution rates with the yn00 method (Yang and Nielsen, 2000) from the PAML package (Yang, 2007). Finally, fast-evolving sequences were identified. Only the protein-coding, intronic or intergenic regions presenting a higher evolutionary rate than the regions most commonly used for evolutionary studies in Fabaceae (rbcL and matK genes, the 5′trnK and trnL introns, and the trnK_trnF, trnL_trnT and trnS_trnG IGSs) and a minimum size of about 300 bp were retained.
A list of all the software programs used in this study, their purpose and availability can be found in the Supplementary Data Table S1.
RESULTS
Organization, gene content and characteristics of the L. luteus plastome
The Lupinus luteus plastome (deposited in GenBank: KC695666) has a length of 151 894 bp, with a quadripartite structure composed of two IRs (25 860 bp) separated by an SSC (17 847 bp) and an LSC (82 327 bp) region (Fig. 2). It contains 111 different genes, including 77 protein-coding genes, 30 tRNA genes and four rRNA genes (Table 1). Protein-coding genes, tRNA and rRNA represent, respectively, 51·6, 1·8 and 6·0% of the plastome. Non-coding DNA, including IGSs and introns, represents 40·6% of the genome. The overall GC content of the L. luteus plastome is 36·6%. It is higher in tRNA and rRNA (53·3 and 55·3%, respectively), slightly higher in protein-coding genes (37·3%), similar in introns (36·3%) and lower in IGSs (30·3%).
Fig. 2.

Circular gene map of the Lupinus luteus (Genistoid; Fabaceae) plastid genome. Genes are represented with boxes inside and outside the first circle to indicate a clockwise or counterclockwise transcription direction, respectively. Genes belonging to different functional groups are colour coded. Read depth coverage of the plastome is represented by proportional radial lines in the inner green circle. The locations of the different main plastomic regions (inverted repeats, large single copy and small single copy) are indicated in the inner circle. The endpoints of the 50-kb and 36-kb inversions are represented by red and green arrows, respectively.
Table 1.
Lupinus luteus plastome characteristics
| Plastome characteristics | |
|---|---|
| Size (bp) | 151 894 |
| LSC size in bp (%) | 82 327 (54·2) |
| SSC size in bp (%) | 17 847 (11·7) |
| IR length in bp (%) | 25 860 (34·1) |
| Size in bp (%) of coding regions | 90 217 (59·4) |
| Size in bp (%) of protein-coding regions | 78 363 (51·6) |
| Size in bp (%) of introns | 19 136 (12·6) |
| Size in bp (%) of rRNA | 9056 (6) |
| Size in bp (%) of tRNA | 2798 (1·8) |
| Size in bp (%) of IGSs | 42 541 (28) |
| No. of different genes | 111 |
| No. of different protein-coding genes | 77 |
| No. of different tRNA genes | 30 |
| No. of different rRNA genes | 4 |
| No. of different genes duplicated by IR | 17 |
| No. of different genes with introns | 18 |
| Overall % GC content* | 36·6 |
| % GC content in protein-coding regions* | 37·3 |
| % GC content in introns* | 36·3 |
| % GC content in IGSs* | 30·3 |
| % GC content in rRNA* | 55·3 |
| % GC content in tRNA* | 53·3 |
*The sequence of the two inverted repeats were taken into account for this analysis.
The L. luteus plastome contains 18 different intron-containing genes (of which six are tRNA), as in most Fabaceae species. All intronic genes contain one intron, apart from two genes (clpP and ycf3) that contain two introns. Within the IR, four rRNA, seven tRNA and five protein-coding genes are repeated. Only the 5′ end of the ycf1 gene (519 bp) is present in the IR, and the gene rps12 is trans-spliced, with the 5′ exon in the LSC and the remaining two exons in the IR.
Thirty different tRNA are present in the L. luteus plastome. They correspond to 28 different codons, at least one for each amino acid. Seven of the 28 different anticodon tRNAs encoded in the Lupinus plastome correspond to the most common codon (where synonymous codons exist). The codon usage is biased towards a high representation of A and T at the third position (Supplementary Data Table S2).
Repeat elements in the L. luteus plastome
All repeat sequences that present a minimum size of 30 bp and with a sequence identity ≥90% were identified in the L. luteus plastome using REPuter (Kurtz et al., 2001). A total of 31 repeats were found (Supplementary Data Table S3), including 13 dispersed short inverted repeats (sIRs), nine palindromes, six dispersed direct repeats and three tandem repeats.
All the palindromic repeats observed in the L. luteus plastome are localized in IGSs (except one in the ycf1 coding sequence) and tandem repeats are mainly found in coding sequences (ycf2).
Most repeats (93·55%) are 30–50 bp long. The largest repeat in the plastome is a 288 bp dispersed direct repeat corresponding to a fragment of ycf2 duplicated in each IR, between rpl23 and tnrI_CAU. This repeated element is absent from extra-Fabales plastomes (C. sativus and P. persica) or from IRLC plastomes in the Papilionoideae, but is present in other non-IRLC Papilionoideae plastomes (G. max, L. japonicus, P. vulgaris and V. radiata), as previously observed by Guo et al. (2007).
Origin of a 36-kb inversion detected in L. luteus
Global alignment and comparison of gene order between the plastomes of L. luteus and other legumes, as well as with outgroups, revealed an inversion of about 36 kb between the trnS-GCU and trnS-GGA genes in L. luteus (Fig. 3). This inversion is highlighted by dot plot analyses that compared the plastome of L. luteus with that of either G. max or C. sativus (Supplementary Data Fig. S1). This unique 36-kb inversion is embedded within the 50-kb inversion that occurred earlier in the Papilionoideae after the divergence between the Cladrastis clade and the rest of the more derived papilionoid legumes (Doyle et al., 1996).
Fig. 3.
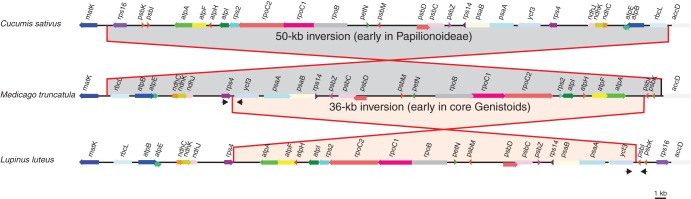
Comparative plastomic maps showing the endpoints of the large 50-kb inversion present in most Papilionideae (Fabaceae) and of a new 36-kb inversion detected in most core Genistoids surveyed in this study. The plastomes of Cucumis sativus, Medicago truncatula and Lupinus luteus were used to represent the structural patterns observed in most flowering plants, in most Papilionoid legumes and in the core Genistoids, respectively. The partial plastomic maps are drawn to scale, and only protein-coding genes are mapped. Approximate positions of diagnostic primer pairs used to detect the presence or absence of the 36-kb inversion are designated by black arrows (not to scale).
To verify the existence of this inversion in Lupinus and to screen other Genistoids and legume species for the presence or absence of this 36-kb inversion, two diagnostic primer pairs were designed. The localization of these primers is indicated in Fig. 3. PCR amplification was expected from the primers located in the rps4 and ycf3 protein-coding sequences only for the species without the inversion, whereas PCR amplification using primers within the ycf3 and psbI genes was only expected in species with the inversion. Since the 36-kb inversion identified in L. luteus is not present in the plastomes available for extra-Fabaceae taxa or in the derived Fabaceae (representative of the Millettoid, Robinoid and IRLC clades), it most probably occurred after the emergence of the Genistoids. For the 11 core Genistoid species screened here (including representatives of the Sophoreae, Thermopsidae and Genisteae tribes), amplifications were only successful when using the diagnostic primers pair for the presence of the inversion. In contrast, all non-core Genistoids tested gave amplification only when using the diagnostic primers pair for the absence of the 36-kb inversion (Fig. 4). In both cases, the results were confirmed by sequencing of the PCR products (deposited in GenBank: KC695667–KC695681) and alignment of the sequences with the homologous plastidic region from other Fabaceae plastomes, including L. luteus.
Fig. 4.

Phylogenetic position of the 36-kb inversion rearrangement (solid black circle) detected in the plastomes of Lupinus and representatives of the core Genistoid clade (Papilionoidae; Fabaceae). All taxa screened by PCR and sequencing for the presence or absence of this inversion have their names labelled in bold. The taxa for which the plastome sequence is publicly available and for which the 36-kb inversion is absent belong to the Millettioids, Robinioids and IRLC (labelled in grey). The phylogenetic tree is redrawn from Cardoso et al. (2012).
Several multiple alignments of the sequences surrounding the endpoints of the 36-kb inversion using Papilionoideae species with or without the 36-kb inversion allowed determination of the exact location of the inversion (Supplementary Data Fig. S2). It occurred between the 3′ end of the trnS-GGA and the trnS-GCU genes that are identical for the last 29 bp and are in inverse orientation (Fig. 5; Supplementary Data Table S3). A similar sIR also exists between trnS-GGA and trnS-UGA that are 9 kb distant.
Fig. 5.

Illustration of the suggested flip–flop recombination event that occurred after the divergence of the Genistoids from the other Fabaceae clades, resulting in a 36-kb inversion (grey region). This recombination event was most probably caused by the presence of inverted repeat sequences (29 bp shown) at the 3′ end of trnS-GGA and trnS-GCU that are 36 kb apart.
Gene content and gene transfers to the nucleus in Lupinus compared with other Fabaceae
The protein-coding gene content of the L. luteus plastome was compared with those of ten other Fabaceae and two outgroup taxa. The aim of this comparison was to examine whether the lupines (representing the core Genistoids) have lost or retained the chloroplast genes known to have lost their functionality in the plastome of various lineages during legume evolution (reviewed in Magee et al., 2010) such as: accD, psaI, rpl22, rpl23, rps16 and ycf4 (as indicated in Fig. 6). Out of these six plastidic genes lost from legume lineages, only rpl22 is missing in the plastome of L. luteus. The functional transfer of this gene to the nucleus, already demonstrated in P. sativum (Gantt et al., 1991), was verified in a lupine species (L. mariae josephi) by the identification of a nuclear rpl22 transcript that is similar to the nuclear rpl22 transcript found in P. sativum. The presence of a chloroplast target peptide was predicted in the L. mariae josephi nuclear rpl22 transcript using a variety of software (data not shown). The alignment and phylogenetic analysis of nuclear and chloroplastic rpl22 sequences (Supplementary Data Fig. S3) showed that the nuclear rpl22 gene observed in Lupinus and Pisum derives from the same transfer event, which occurred in the common ancestor of all flowering plants (Gantt et al., 1991). Concerning the other chloroplast genes lost during Fabaceae evolution, investigations were performed to determine whether they could be at an intermediate stage of functional transfer to the nucleus. We identified nuclear rps16 transcripts in L. mariae josephi that were similar to the M. truncatula nuclear rps16 genes (Ueda et al., 2008), but no nuclear accD, psaI, rpl23 or ycf4 transcripts could be detected.
Fig. 6.

Chloroplast genes and introns lost during Fabaceae evolution. All Fabaceae species whose plastome has been fully sequenced are presented in the phylogenetic tree (redrawn from Cardoso et al., 2012). Black circles on branches indicate intron losses, whereas triangles show the genes recently lost from the plastome in various Fabaceae species. Black triangles indicate genes that were shown to be functionally transferred to the nucleus (Gantt et al., 1991; Magee et al., 2010), including the transfer of rpl22 in Lupinus luteus detected in this study. The grey triangle indicates the functional replacement of the plastomic rps16 gene by a mitochondrial gene functionally transferred to the nucleus (Ueda et al., 2008). White triangles show the plastid genes lost during Fabaceae evolution and for which no functional replacement in the nucleus has been observed. The independent losses of ycf4 in Lathyrus sativus and Pisum sativum were inferred from the results of Magee et al. (2010) who showed the presence of intact ycf4 genes in some Lathyrus species.
Sequence divergence between the plastome of L. luteus and other Fabaceae
A comparison of pairwise distances (K2p) calculated for non-coding regions between L. luteus and other legumes (Supplementary Data Table S4) revealed that, as expected, IGSs evolve significantly more rapidly than introns. The slowest evolving regions are tRNAs and rRNAs (Supplementary Data Table S4). For introns (Fig. 7A), the mean sequence divergence ranged from 0·028 (for the rps12 intron) to 0·270 (for clpP intron1). The two main introns previously used for phylogenetic inference in legumes showed relatively low rates of variation: 0·100 for the trnL intron (501 bp length) and 0·148 for the trnK 5′ intron (318 bp length). Among introns, seven exhibited a higher level of divergence (Fig. 7A): the trnG-UCC intron (K2p = 0·183; 698 bp), rpoC1 intron (0·164; 766 bp), clpP intron 2 (0·230; 739 bp), clpP intron 1 (0·270; 655 bp), petD intron (0·224; 743 bp), rpl16 intron (0·195; 1155 bp) and ndhA intron (0·215; 1171 bp). The highest mean sequence divergence of IGS regions corresponds to the accD_psaI region (0·473). Among IGSs and in comparison with the IGS regions most used for legume phylogeny (trnL_trnF, mean K2p = 0·255; trnL_trnT, 0·352; trnS_trnG, 0·316), five IGSs >300 bp showed divergence rates slightly or significantly higher than trnL_trnT, i.e. ycf4_cemA (0·357; 317 bp), rpl36_rps8 (0·357; 453), psbZ_trnG-GCC (0·357; 345 bp), trnV-UAC_ndhC (0·355; 497 bp) and accD_psaI (0·473; 293 bp (Fig. 7A). For protein-coding regions (Fig. 7B; Supplementary Data Table S5), the evolutionary rates have been evaluated by comparison of their synonymous (Ks) nucleotide substitution rates (Fig. 7B). The mean divergence rate between L. luteus and the other legume genes ranged fom 0·072 (for rpl23; 282 bp) to 0·667 (for rps16; 47 bp), with most loci presenting mean Ks values lower than those of the two protein-coding genes used for phylogenetic inference in legumes, matK (0·235; 1521 bp) and rbcL (0·367; 1428 bp). Fourteen genes displayed Ks values higher than rbcL, of which nine are >300 bp: rpoC2 (0·419; 4149 bp), rps16 (0·667; 407 bp), accD (0·538; 1497 bp), ycf4 (0·659; 555 bp), rps8 (0·388; 405 bp), rpl14 (0·441; 369 bp), ycf1 (0·518; 5296 bp), ndhH (0·394; 1182) and ndhF (0·454; 2241 bp). For most loci (65/77), the non-synonymous nucleotide substitution (Ka) values calculated between Lupinus and the other legumes were <0·1 (Supple-mentary Data Table S6). Among the 12 remaining loci, only five displayed higher values than the reference matK gene (mean Ka = 0·132): rpl32 (0·147), rps16 (0·169), accD (0·184) and particularly ycf1 (0·306) and ycf4 (0·398). Regarding these low values of Ka, the Ka/Ks ratio calculated for each protein-coding gene (Supplementary Data Table S7) was <1 and even <0·5 for almost all loci, indicating that plastidic genes are under a high negative (i.e. purifying) selective constraint (Kimura, 1977; Messier and Stewart, 1997). Nevertheless, it can be noted that the three ycf genes (ycf1, ycf2 and ycf4) exhibit remarkably higher Ka/Ks values (0·601, 0·682 and 0·649, respectively) than all the other genes, indicating an increase in their sequence evolutionary rate.
Fig. 7.

Mean sequence divergence ± s.e. between homologous regions of the Lupinus luteus and other legume plastomes. The x-axis lists intronic, intergenic and protein-coding regions in the same order as in the L. luteus plastome. (A) Red and blue filled circles show mean sequence divergence for each orthologous intronic or intergenic pair, calculated using the K2p model (Kimura, 1980). (B) Green filled squares show mean sequence divergence for each orthologous protein-coding gene pair, estimated with the synonymous mutation rate (Ks) and using the yn00 method (Yang, 2007). The various regions (intronic, intergenic or protein-coding gene pair) previously used in Fabaceae phylogenetic studies are indicated with red, blue and green arrows, respectively. The intronic, intergenic or protein-coding regions presenting a higher evolutionary rate than those previously used in Fabaceae evolutionary studies and presenting a minimum size of 300 bp are indicated with red, blue or green asterisks, respectively.
Altogether, these analyses allow circumscription of fast-evolving regions in the legume plastomes, as inferred from pairwise comparison of Lupinus with the other available legume plastomes (highlighted in Supplementary Data Fig. S4). Among these regions, three are remarkable: one in the SSC, between ycf1 and the ndhA intron (9043 bp); two in the LSC, around the rpl36-rpl16 genes (3178 bp), and the accD-ycf4_cemA region (2968 bp) that exhibits the highest rates of sequence divergence for genes (rps16, accD and ycf4) and IGSs (accD_psaI and ycf4_cemA). The latter region, which includes the ycf4 gene, was shown to have a dramatic increase in its evolutionaty rate in the NPAAA clade (including Millettioids, Robinioids, and IRLC) and most particularly in Lathyrus (Magee et al., 2010). To investigate whether such acceleration also occurred in the lupine lineage, maximum likelihood phylogenetic analyses using legume ycf4 gene sequences (including L. luteus ycf4) and based on Ks and Ka substitution rates were performed. Our results (Supplementary Data Fig. S5) do not provide evidence of such acceleration in the ycf4 gene in Lupinus, in accordance with the previous results obtained by Magee et al. (2010) using a few Genistoid members (Crotalaria, Goodia and Laburnum).
The other regions showing peaks of divergence when comparing Lupinus with other legume plastomes include some isolated genes (rpoC2 and ndhF), introns (trnG-UCC, rpoC1 and petD) and IGSs (trnV_ndhC and psbZ_trnG-GCC) that were not previously detected as fast-evolving regions in NPAAA clade members.
DISCUSSION
In this work the plastome of Lupinus luteus has been sequenced using NGS technologies. Its size and gene content are within the range found in plastomes containing two IRs (Raubeson and Jansen, 2005). It is AT rich (with the exception of rRNA and tRNA genes) and the codon usage is biased toward a high representation of A and T at the third position as previously observed by Clegg et al. (1994). This sequence, which represents the first plastome sequenced in the core Genistoids, is of major interest because all legume plastomes sequenced so far belong to only three Papilionoid clades, the Millettioids, the Robinioids and the IRLC, which derive from within the NPAAA clade (Cardoso et al., 2012). Thus it was essential to sequence plastomes from representatives of other Papilionoid lineages in order to have a better understanding of the unusual plastome evolution observed in legumes (Jansen and Ruhlman, 2012). Most photosynthetic angiosperms have a highly conserved plastome organization, except the Campanulaceae, Fabaceae and Geraniaceae families that exhibit remarkable and extensive rearrangements (Jansen and Ruhlman, 2012). Within the Fabaceae, one of the most remarkable inversions that occurred after the emergence of the family is the 50-kb inversion, which is shared by most papilionoid taxa (Doyle et al., 1996). The plastome sequences of three IRLC species, L. sativus (Magee et al., 2010), P. sativum (Palmer and Herbon, 1988) and T. subterraneum (Cai et al., 2008), revealed that, relative to the ancestral angiosperm plastome organization, and after the 50-kb inversion event, six, eight and fifteen inversions occurred, respectively (Magee et al., 2010). Within the 50-kb clade, our study reveals that the L. luteus plastome experienced an additional 36-kb inversion internal to the 50-kb inversion, which most probably occurred at the origin of the core Genistoids. Previous molecular characterization of large plastomic inversion endpoints in a few plant families or genera, including the 50-kb inversion present in most Papilionoideae (Doyle et al., 1996), the 22-kb inversion in Asteraceae (Kim et al., 2005), the 42-kb inversion in Abies (Tsumura et al., 2000) or the 21-kb inversion in Jasminae (Lee et al., 2007), showed that these large plastomic inversions were often associated with sIRs present within, or adjacent to, a tRNA. The detailed survey of the regions surrounding the 36-kb inversion endpoints in core Genistoids allowed us to determine that this inversion is most likely to be due to the presence of sIR motifs (29 identical nucleotides) at the 3′ end of trnS-GGA and trnS-GCU. The role of repeated elements present in inverse orientation in promoting flip–flop recombination resulting in inversions has been previously demonstrated using tobacco transplastomic lines (Rogalski et al., 2006). Such repeated elements can promote plastid DNA inversions which may vary in size from a few base pairs to several kilobases (reviewed in Downie and Palmer, 1992). Minor inversions are more common than major ones and mainly occur in non-coding regions, IGSs and introns (Palmer, 1985). Interestingly the sIR motif in trnS-GGA and trnS-GCU, which caused the 36-kb inversion in the core Genistoids, is present in almost all Rosids (Supplementary Data Table S8) and is separated by at least 30 kb. Thus, even though this 36-kb inversion was only observed in the core Genistoids, it could have occurred in any other rosid species.
The L. luteus plastome contains fewer repeats (31) than other Fabaceae species, such as V. radiata, L. japonicus, G. max or M. truncatula that have 50, 67, 104 and 191 repeats, respectively (Saski et al., 2005; Tangphatsornruang et al., 2010). Most L. luteus repeats are relatively small in size (90% are 30–50 bp in size). The longest repeat observed in L. luteus is a 288 bp direct repeat (within the IR) that is also present in the non-IRLC Papilionidae (G. max, L. japonicus, P. vulgaris and V. radiata) but not in the IRLC or outgroup taxa (C. sativus and P. persica). The low number of repeats observed in Lupinus is in stark constrast to the T. subterraneum plastome that contains a high number of large repeats and shows a high rate of rearrangement: 14 inversions occurred since its divergence with other IRLC species (Cai et al., 2008; Magee et al., 2010). The number of large repeats was demonstrated to be positively correlated to the degree of plastome rearragements in plants (Maul et al., 2002; Pombert et al., 2005; Guisinger et al., 2011). Within the repeats observed in Lupinus, 42% (13/31) are dispersed sIRs (6–66 kb distant) that could promote inversions. However, apart from the dispersed sIR at the origin of the 36-kb inversion in the core Genistoid and another repeat between trnS-UGA and the trnS-GGA (9 kb distant), all the others would presumably lead to the loss of functionality of one or several genes in the case of an inversion event and thus may be deleterious (Ruf et al., 1997: Drescher et al., 2000). Whilst the above 9-kb region is potentially prone to inversion, to date no evidence of such an event has been observed from this or previous studies in other Fabaceae species.
The rarity of plastomic rearrangements in flowering plants makes these characters powerful phylogenetic markers (Kim et al., 2005) since they present an extremely low level of homoplasy (Cosner et al., 2004). The 36-kb inversion identified in this study is present in all core Genistoid species surveyed (12) and therefore provides a robust additional synapomorphy supporting monophyly of the core Genistoids (Crisp et al., 2000). Further screening of representatives from Brongniartieae and Bowdichia clades, shown to be closely related to the core Genistoids (Cardoso et al., 2012), will determine whether this 36-kb inversion is strictly specific to the core Genistoids or whether it occurred earlier or at the base of the large Genistoid s.l. assemblage (includes Brongniartieae and Bowdichia clades). Thus, after the 50-kb inversion that is shared by a majority of Papilionideae (Doyle et al., 1996), and the 78-kb inversion supporting the Papilionoid subtribe Phaseolinae (Bruneau et al., 1990) in legumes, this 36-kb inversion represents an additional example highlighting the phylogenetic usefulness of plastidic inversions. Such clade-demarcating inversions were also detected in other Angiosperm families. Within the Asteraceae, a 22-kb inversion allowed identification of the subtribe Barnadesiinae as the most primitive lineage in the family (Jansen and Palmer, 1987). In the Campanulaceae, which also have highly rearranged plastomes, reliable phylogenetic relationships could be reconstructed within the family only based on the use of the numerous rearrangements (including inversions) as characters (Cosner et al., 2004). Interestingly, there is also evidence of specific mutational and restructuring events that affected the nuclear genome of Lupinus (Mahé et al., 2011a), which suggests that the Genistoids experienced noteworthy genomic changes, in both the plastid and the nuclear genomes, after their divergence from the NPAAA papilionoid lineages (approx. 50–56 million years ago).
Gene content is highly conserved among photosynthetic angiosperm plastomes (Timmis et al., 2004). However, within the Fabaceae, several chloroplastic genes (accD, psaI, rpl22, rpl23, rps16 and ycf4) have been lost recently and independently in various lineages (Magee et al., 2010). However, within the L. luteus plastome, only the rpl22 gene is missing, which is in accordance with the previous finding of Gantt et al. (1991) who demonstrated that the functional transfer of this gene from the chloroplast to the nucleus occurred in a common ancestor of all flowering plants, and thus preceded its loss from the chloroplast genome by about 100 million years (Supplementary Data Fig. S3). Among the chloroplast genes that have been lost in legume lineages following their divergence from the common ancestor with lupines, we found that the rps16 gene is at an intermediate stage of functional replacement in the Lupinus nuclear genome, as it is still represented by a functional copy in the chloroplast genome while another is in the nucleus. This nuclear-encoded rps16 gene targeted to the plastid is of mitochondrial origin and was transferred prior to the monocot–dicot divergence (Ueda et al., 2008).
The evaluation of sequence divergence between Lupinus and the other sequenced legumes allowed identification of fast-evolving sequences (Fig. 7; Supplementary Data Fig. S4). This information is essential for a better understanding of the dynamic nature of plastome evolution in legumes and for improving legume phylogeny, especially within the Genistoids and the genus Lupinus (Eastwood et al., 2008; Mahé et al., 2011a; Cardoso et al., 2012). As expected, most coding regions are well conserved, particularly in the IR region, and in most cases IGSs are evolving faster than introns, in accordance with previous observations (Clegg and Zurawski, 1991; Raubeson et al., 2007). Compared with the plastid sequences used in legume evolutionary studies, we have detected several sequences (Fig. 7) that exhibit higher rates of divergence: (1) seven introns (trnG-UCC, rpoC1, rpl16, ndhA, petD and clpP introns 1 and 2); (2) five IGSs (ycf4_cemA, rpl36_rps8, psbZ_trnG-GCC, trnV-UAC_ndhC and accD_psaI); and (3) eight protein-coding genes (rpoC2, accD, ycf4, rps8, rpl14, ycf1, ndhH and ndhF). Interestingly, most of these variable regions have not been or have rarely been employed in legume phylogeny [e.g. the trnS-trnG region in Lupinus by Drummond (2008); ycf1 in Astragalus by Bartha et al. (2013)], and thus represent a new set of markers to explore evolutionary relationships within legumes. Each of these sequences needs to be tested in order to evaluate at which taxonomic level and in which lineages they could be more informative and useful. As an example, the remarkable increase in the evolutionary rate observed in the ycf4 gene is specific to the IRLC, Robinioid and Millettioid lineages (NPAAA clade), and occurred after the divergence of the latter from the other legumes (Magee et al., 2010). Thus, this region is most probably a good candidate for the NPAAA clade but seems less interesting for phylogenetic inference within the Genistoids and earlier legume lineages. In contrast, we have detected several loci exhibiting an increase in their evolutionary rate that is specific to the lupine/genistoid lineage. These loci include the rpoC2 and ndhF genes, the trnG-UCC, rpoC1 and petD introns, and the trnV_ndhC and psbZ_trnG-GCC IGSs. Regardless of the specificity and degree of utility of each locus, altogether these variable sequences constitute an important source of novel characters for single- or multigene-based reconstruction of evolutionary patterns in legumes at various taxonomic levels.
These variable sequences (mentioned above) are distributed in well-circumscribed fast-evolving regions that shape the legume plastome landscape (Fig. 7; Supplementary Data Fig. S4). Interestingly, three of these variable regions are located at boundaries of the 50-kb inversion (rps16_ycf4 region), the 36-kb inversion (trnS-GCU_trnG-UCC region) and the IR region (ycf1 region) (Fig. 7). As previously pointed out by Magee et al. (2010), these fast-evolving regions include gene and intron losses, such as genes lost from the rps16_ycf4 region (rps16, accD, psaI, rpl23 and ycf4) and introns lost from the clpP_rps12 and the rpl16 regions (Fig. 7). This suggests that these regions are most probably involved in structural rearrangements and thus represent unstable regions or hotspots that contribute significantly to the evolutionary dynamics of legume plastomes. Future research on the efficiency of the four classes of nuclear-encoded genes that are involved in chloroplast DNA repair and the maintenance of plastome stability (Maréchal and Brisson, 2010; Guisinger et al., 2011) may reveal whether one or several of these four genes are implicated in legume plastome evolution. Additionally, this study demonstrates that it is essential to sequence plastomes from other papilionoid and earlier legume lineages that remain unexplored to date in order to have a better understanding of the atypical plastome evolution observed in this family.
SUPPLEMENTARY DATA
ACKNOWLEDGEMENTS
We thank Professor J. N. Timmis (University of Adelaide) for his critical reading of the manuscript. This work was supported by UMR-CNRS Ecobio (Rennes, France), and benefited from facilities and support from the ‘Plate-forme Génomique Environnementale et Fonctionnelle’ (OSUR: INEE-CNRS) and the Genouest Bioinformatic Plateform (University of Rennes 1). This work was supported by the Région Bretagne and the European Union Seventh Framework Programme [FP7-CIG-2013–2017; grant no. 333709 to M. R.-G.]. We are grateful to the reviewers for helpful comments and suggestions to improve the early draft of the manuscript.
LITTERATURE CITED
- Ainouche A, Bayer RJ. Phylogenetic relationships in Lupinus (Fabaceae: Papilionoideae) based on internal transcribed spacer sequences (ITS) of nuclear ribosomal DNA. American Journal of Botany. 1999;86:590–607. [PubMed] [Google Scholar]
- Ainouche A, Bayer RJ, Misset MT. Molecular phylogeny, diversification and character evolution in Lupinus (Fabaceae) with special attention to Mediterranean and African lupines. Plant Systematics and Evolution. 2004;246:211–222. [Google Scholar]
- Bartha L, Dragos N, Molnár V, Sramkó G. Molecular evidence for reticulate speciation in Astragalus (Fabaceae) as revealed by a case study from sect. Dissitiflori. Botany. 2013;91:702–714. [Google Scholar]
- Bruneau A, Doyle JJ, Palmer JD. A chloroplast DNA structural mutation as a subtribal character in the Phaseoleae (Leguminosae) Systematic Botany. 1990;15:378–386. [Google Scholar]
- Cai Z, Guisinger M, Kim HG, et al. Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. Journal of Molecular Evolution. 2008;67:696–704. doi: 10.1007/s00239-008-9180-7. [DOI] [PubMed] [Google Scholar]
- Cardoso D, de Queiroz LP, Pennington RT, et al. Revisiting the phylogeny of papilionoid legumes: new insights from comprehensively sampled early-branching lineages. American Journal of Botany. 2012;99:1991–2013. doi: 10.3732/ajb.1200380. [DOI] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, Suhai S. Genome sequence assembly using trace signals and additional sequence information; Computer Science and Biology; Germany: GCB, Hannover; 1999. pp. 45–56. Proceedings of the German Conference on Bioinformatics, GCB '99. [Google Scholar]
- Clegg M, Zurawski G. Chloroplast DNA and the study of plant phylogeny: present status and future prospects. In: Soltis PS, Soltis DE, Doyle JJ, editors. Molecular systematics of plants. New York: Chapman & Hall; 1991. pp. 1–13. [Google Scholar]
- Clegg MT, Gaut BS, Learn GH, Jr, Morton BR. Rates and patterns of chloroplast DNA evolution. Proceedings of the National Academy of Sciences, USA. 1994;91:6795–6801. doi: 10.1073/pnas.91.15.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosner ME, Raubeson LA, Jansen RK. Chloroplast DNA rearrangements in Campanulaceae: phylogenetic utility of highly rearranged genomes. BMC Evolutionary Biology. 2004;4:27. doi: 10.1186/1471-2148-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M, Gilmore S, van Wyk B. Molecular phylogeny of the Genistoid tribes of Papilionoid legumes. London: Royal Botanic Gardens, Kew; 2000. [Google Scholar]
- Cronk Q, Ojeda I, Pennington RT. Legume comparative genomics: progress in phylogenetics and phylogenomics. Current Opinion in Plant Biology. 2006;9:99–103. doi: 10.1016/j.pbi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Cui L, Veeraraghavan N, Richter A, et al. ChloroplastDB: the Chloroplast Genome Database. Nucleic Acids Research. 2006;34:692–696. doi: 10.1093/nar/gkj055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie SR, Palmer JD. Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In: Soltis PS, Soltis DE, Doyle JJ, editors. Molecular systematics of plants. New York: Chapman & Hall; 1992. pp. 14–35. [Google Scholar]
- Doyle JJ, Doyle JL, Ballenger JA, Palmer JD. The distribution and phylogenetic significance of a 50-kb chloroplast DNA inversion in the flowering plant family Leguminosae. Molecular Phylogenetics and Evolution. 1996;5:429–438. doi: 10.1006/mpev.1996.0038. [DOI] [PubMed] [Google Scholar]
- Drescher A, Ruf S, Calsa T, Jr, Carrer H, Bock R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. The Plant Journal. 2000;22:97–104. doi: 10.1046/j.1365-313x.2000.00722.x. [DOI] [PubMed] [Google Scholar]
- Drummond CS. Diversification of Lupinus (Leguminosae) in the western New World: derived evolution of perennial life history and colonization of montane habitats. Molecular Phylogenetics and Evolution. 2008;48:408–421. doi: 10.1016/j.ympev.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Eastwood RJ, Drummond CS, Schifino-Wittmann MT, Hughes CE. Australia: Fremantle; 2008. Diversity and evolutionary history of lupins – insights from new phylogenies. Proceedings of the 12th International Lupin Conference – Lupins for Health and Wealth. [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. Journal of Molecular Biology. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence-limits on phylogenies – an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO Journal. 1991;10:3073–3078. doi: 10.1002/j.1460-2075.1991.tb07859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcinuno RM, Fernandez-Hernando P, Camara C. Evaluation of pesticide uptake by Lupinus seeds. Water Research. 2003;37:3481–3489. doi: 10.1016/S0043-1354(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Guillon F, Champ MM. Carbohydrate fractions of legumes: uses in human nutrition and potential for health. British Journal of Nutrition. 2002;88:293–306. doi: 10.1079/BJN2002720. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Molecular Biology and Evolution. 2011;28:583–600. doi: 10.1093/molbev/msq229. [DOI] [PubMed] [Google Scholar]
- Guo X, Castillo-Ramirez S, Gonzalez V, et al. Rapid evolutionary change of common bean (Phaseolus vulgaris L) plastome, and the genomic diversification of legume chloroplasts. BMC Genomics. 2007;8:228. doi: 10.1186/1471-2164-8-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C, Eastwood R. Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proceedings of the National Academy of Sciences; USA. 2006. pp. 10334–10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RK, Palmer JD. A chloroplast DNA inversion marks an ancient evolutionary split in the sunflower family (Asteraceae). Proceedings of the National Academy of Sciences; USA. 1987. pp. 5818–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RK, Ruhlman TA. Plastid genomes of seed plants. In: Bock R, Knoop V, editors. Genomics of chloroplast and mitochondria. New York: Springer; 2012. pp. 103–126. [Google Scholar]
- Jansen RK, Cai Z, Raubeson LA, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proceedings of the National Academy of Sciences; USA. 2007. pp. 19369–19374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RK, Wojciechowski MF, Sanniyasi E, Lee SB, Daniell H. Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae) Molecular Phylogenetics and Evolution. 2008;48:1204–1217. doi: 10.1016/j.ympev.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Kaneko T, Sato S, Nakamura Y, Tabata S. Complete structure of the chloroplast genome of a legume, Lotus japonicus. DNA Research. 2000;7:323–330. doi: 10.1093/dnares/7.6.323. [DOI] [PubMed] [Google Scholar]
- Kazakoff SH, Imelfort M, Edwards D, et al. Capturing the biofuel wellhead and powerhouse: the chloroplast and mitochondrial genomes of the leguminous feedstock tree Pongamia pinnata. PLoS One. 2012;7:e51687. doi: 10.1371/journal.pone.0051687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Choi KS, Jansen RK. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae) Molecular Biology and Evolution. 2005;22:1783–1792. doi: 10.1093/molbev/msi174. [DOI] [PubMed] [Google Scholar]
- Kimura M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature. 1977;267:275–276. doi: 10.1038/267275a0. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23:1026–1028. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Research. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Reasearch. 2001;29:4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin M, Doyle JJ, Palmer JD. Evolutionary significance of the loss of the chloroplast –DNA inverted repeat in the Leguminosae subfamily Papilionoideae. Evolution. 1990;44:390–402. doi: 10.1111/j.1558-5646.1990.tb05207.x. [DOI] [PubMed] [Google Scholar]
- Lee HL, Jansen RK, Chumley TW, Kim KJ. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Molecular Biology and Evolution. 2007;24:1161–1180. doi: 10.1093/molbev/msm036. [DOI] [PubMed] [Google Scholar]
- Lewis G, Schrire B, MacKinder B, Lock M. Legumes of the world. London: Royal Botanical Gardens, Kew; 2005. [Google Scholar]
- Magee AM, Aspinall S, Rice DW, et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Research. 2010;20:1700–1710. doi: 10.1101/gr.111955.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni C, Sessa F, Accardo E, et al. Conglutin gamma, a lupin seed protein, binds insulin in vitro and reduces plasma glucose levels of hyperglycemic rats. Journal of Nutritional Biochemistry. 2004;15:646–650. doi: 10.1016/j.jnutbio.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Mahé F, Markova D, Pasquet R, Misset MT, Ainouche A. Isolation, phylogeny and evolution of the SymRK gene in the legume genus Lupinus L. Molecular Phylogenetics and Evolution. 2011a;60:49–61. doi: 10.1016/j.ympev.2011.04.017. [DOI] [PubMed] [Google Scholar]
- Mahé F, Pascual H, Coriton O, et al. New data and phylogenetic placement of the enigmatic old world lupin: Lupinus mariae-josephi H. Pascual. Genetic Resources and Crop Evolution. 2011b;58:101–114. [Google Scholar]
- Maul JE, Lilly JW, Cui L, et al. The Chlamydomonas reinhardtii plastid chromosome: islands of genes in a sea of repeats. The Plant Cell. 2002;14:2659–2679. doi: 10.1105/tpc.006155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A, Brisson N. Recombination and the maintenance of plant organelle genome stability. New Phytologist. 2010;186:299–317. doi: 10.1111/j.1469-8137.2010.03195.x. [DOI] [PubMed] [Google Scholar]
- Messier W, Stewart C-B. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- Millen RS, Olmstead RG, Adams KL, et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. The Plant Cell. 2001;13:645–658. doi: 10.1105/tpc.13.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Bell CD, Soltis PS, Soltis DE. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proceedings of the National Academy of Sciences; USA. 2007. pp. 19363–19368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proceedings of the National Academy of Sciences; USA. 2010. pp. 4623–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JD. Comparative organization of chloroplast genomes. Annual Review of Genetics. 1985;19:325–354. doi: 10.1146/annurev.ge.19.120185.001545. [DOI] [PubMed] [Google Scholar]
- Palmer JD, Herbon LA. Plant mitochondrial-DNA evolves rapidly in structure, but slowly in sequence. Journal of Molecular Evolution. 1988;28:87–97. doi: 10.1007/BF02143500. [DOI] [PubMed] [Google Scholar]
- Palmer JD, Thompson WF. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell. 1982;29:537–50. doi: 10.1016/0092-8674(82)90170-2. [DOI] [PubMed] [Google Scholar]
- Paradis E, Bolker B, Claude J, et al. Package ‘ape’. 2011. Available at: http://cran.r-project.org/web/packages/ape/ape.pdf .
- Perry AS, Brennan S, Murphy DJ, Kavanagh TA, Wolfe KH. Evolutionary re-organisation of a large operon in adzuki bean chloroplast DNA caused by inverted repeat movement. DNA Research. 2002;9:157–162. doi: 10.1093/dnares/9.5.157. [DOI] [PubMed] [Google Scholar]
- Pierleoni A, Martelli PL, Fariselli P, Casadio R. BaCelLo: a balanced subcellular localization predictor. Bioinformatics. 2006;22:e408–e416. doi: 10.1093/bioinformatics/btl222. [DOI] [PubMed] [Google Scholar]
- Pilvi TK, Jauhiainen T, Cheng ZJ, Mervaala EM, Vapaatalo H, Korpela R. Lupin protein attenuates the development of hypertension and normalises the vascular function of NaCl-loaded Goto-Kakizaki rats. Journal of Physiology and Pharmacology. 2006;57:167–176. [PubMed] [Google Scholar]
- Pombert JF, Otis C, Lemieux C, Turmel M. The chloroplast genome sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) reveals unusual structural features and new insights into the branching order of chlorophyte lineages. Molecular Biology and Evolution. 2005;22:1903–1918. doi: 10.1093/molbev/msi182. [DOI] [PubMed] [Google Scholar]
- Raubeson LA, Jansen RK. Chloroplast genomes of plants. Wallingford, UK: CABI Publishing; 2005. [Google Scholar]
- Raubeson LA, Peery R, Chumley TW, et al. Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics. 2007;8:174. doi: 10.1186/1471-2164-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski M, Ruf S, Bock R. Tobacco plastid ribosomal protein S18 is essential for cell survival. Nucleic Acids Research. 2006;34:4537–4545. doi: 10.1093/nar/gkl634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf S, Kössel H, Bock R. Targeted inactivation of a tobacco intron-containing open reading frame reveals a novel chloroplast-encoded photosystem I-related gene. Journal of Cell Biology. 1997;139:95–102. doi: 10.1083/jcb.139.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor–joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Saski C, Lee SB, Daniell H, et al. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Molecular Biology. 2005;59:309–322. doi: 10.1007/s11103-005-8882-0. [DOI] [PubMed] [Google Scholar]
- Small I, Peeters N, Legeai F, Lurin C. Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics. 2004;4:1581–1590. doi: 10.1002/pmic.200300776. [DOI] [PubMed] [Google Scholar]
- Sprent JI. Evolving ideas of legume evolution and diversity: a taxonomic perspective on the occurrence of nodulation. New Phytologist. 2007;174:11–25. doi: 10.1111/j.1469-8137.2007.02015.x. [DOI] [PubMed] [Google Scholar]
- Stefanovic S, Pfeil BE, Palmer JD, Doyle JJ. Relationships among phaseolid legumes based on sequences from eight chloroplast regions. Systematic Botany. 2009;34:115–128. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangphatsornruang S, Sangsrakru D, Chanprasert J, et al. The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA Research. 2010;17:11–22. doi: 10.1093/dnares/dsp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmis JN, Ayliffe MA, Huang CY, Martin W. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nature Review Genetics. 2004;5:123–135. doi: 10.1038/nrg1271. [DOI] [PubMed] [Google Scholar]
- Tsumura Y, Suyama Y, Yoshimura K. Chloroplast DNA inversion polymorphism in populations of Abies and Tsuga. Molecular Biology and Evolution. 2000;17:1302–1312. doi: 10.1093/oxfordjournals.molbev.a026414. [DOI] [PubMed] [Google Scholar]
- Ueda M, Fujimoto M, Arimura SI, Tsutsumi N, Kadowaki KI. Presence of a latent mitochondrial targeting signal in gene on mitochondrial genome. Molecular Biology and Evolution. 2008;25:1791–1793. doi: 10.1093/molbev/msn139. [DOI] [PubMed] [Google Scholar]
- Wojciechowski MF, Lavin M, Sanderson MJ. A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. American Journal of Botany. 2004;91:1846–1862. doi: 10.3732/ajb.91.11.1846. [DOI] [PubMed] [Google Scholar]
- Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Molecular Biology and Evolution. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.