

Abstract
Activation of phosphatidylinositol 3‐kinase (PI3K) signaling is involved in carcinogenesis and cancer progression. The PI3K inhibitors are considered candidate drugs for cancer treatment. Here, we describe a drug screening system for novel PI3K inhibitors using Saccharomyces cerevisiae strains with deleterious mutations in the ATP‐binding cassette transporter genes, because wild‐type S. cerevisiae uses drug efflux pumps for reducing intracellular drug concentrations. By screening the chemical library of the Screening Committee of Anticancer Drugs, we identified the histone deacetylase (HDAC) inhibitor romidepsin (FK228) and its novel analogs. In vitro PI3K activity assays confirmed that these compounds directly inhibit PI3K activity at μM‐range concentrations. FK‐A5 analog was the most potent inhibitor. Western blotting revealed that these compounds inhibit phosphorylation of protein kinase B and downstream signaling components. Molecular modeling of the PI3K–FK228 complex indicated that FK228 binds to the ATP‐binding pocket of PI3K. At μM‐range concentrations, FK228 and FK‐A5 show potent cytotoxicity, inducing apoptosis even in HDAC inhibitor‐resistant cells. Furthermore, HDAC/PI3K dual inhibition by FK228 and FK‐A5 at μM‐range concentrations potentiates the apoptosis induction, mimicking the effect of combining specific HDAC and PI3K inhibitors. In this study, we showed that FK228 and its analogs directly inhibit PI3K activity and induce apoptosis at μM‐range concentrations, similar to HDAC/PI3K dual inhibition. In future, optimizing the potency of FK228 and its analogs against PI3K may contribute to the development of novel HDAC/PI3K dual inhibitors for cancer treatment.
Phosphatidylinositol 3‐kinase (PI3K) phosphorylates phosphatidylinositol 4,5‐bisphosphate (PIP2) at the 3‐OH group of the inositol ring, generating phosphatidylinositol 3,4,5‐trisphosphate (PIP3).1 In turn, PIP3 activates protein kinase B (AKT) and downstream molecules, leading to increased cell growth, proliferation, and survival.2 Class IA PI3K is composed of a catalytic subunit, p110, and a regulatory subunit, p85. PIK3CA encodes p110α, and is frequently mutated in human cancers.3 Because almost all of the mutations are functionally active,4 activation of PI3K is likely to play a crucial role in carcinogenesis and cancer progression. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) dephosphorylates PIP3 to PIP2 as the catalytic counterpart of PI3K.5 In addition to PIK3CA mutations, inactivating mutations in, or loss of, PTEN have also been observed in human cancers, supporting the involvement of PI3K in cancer.6 Therefore, inhibitors of PI3K are considered to be candidate drugs for cancer therapy. Indeed, some PI3K inhibitors have entered clinical trials, but efforts are still underway to develop new PI3K inhibitors.7
To discover novel PI3K inhibitors, we took advantage of a drug screening system that uses a strain of the yeast Saccharomyces cerevisiae with deleterious mutations in ATP‐binding cassette (ABC) transporter genes. We used this strain because intracellular drug concentrations in yeast with wild‐type ABC transporter genes are efficiently reduced by drug efflux pumps.8 The screening system is based on growth inhibition induced by overexpression of membrane‐localized p110α in yeast, in which no endogenous p110 homolog is expressed.9 Conversion of the essential PIP2 pool to PIP3 by exogenous p110α expression impairs yeast growth by altering morphogenesis and vesicular trafficking.10 The PI3K inhibitor LY294002 is reportedly able to reverse this p110α‐induced growth inhibition.10 Similarly, other compounds with the ability to inhibit PI3K can rescue cells from p110α‐induced growth inhibition. Using this system, we screened the deposited chemical library of the Screening Committee of Anticancer Drugs (SCADS) and thereby isolated a histone deacetylase (HDAC) inhibitor, romidepsin (FK228), and its analogs. FK228 is a potent HDAC inhibitor, with recent approval for the treatment of cutaneous T‐cell lymphoma.11 The HDAC inhibitors have pleiotropic antitumor activities, because of their ability to act on non‐histone targets.12 However, there is no evidence that HDAC inhibitors have kinase inhibitory activities. Here, we showed that FK228 and its analogs directly inhibit PI3K activity, and we evaluated their cytotoxicity as HDAC/PI3K dual inhibitors.
Materials and Methods
Plasmids
The plasmid pSJ01 is a CEN/ARS URA3 vector, which expresses p110α plus the palmitoylation signal from H‐Ras (p110α–CAAX) under the control of GAL1 promoter. To generate pSJ01, the full‐length open reading frame PIK3CA cDNA (a gift from Peter K. Vogt, The Scripps Research Institute, La Jolla, CA, USA) was amplified by PCR adding the CAAX motif immediately before the stop codon. The plasmid pSJ21 is a CEN/ARS LEU2 vector, which expresses wild‐type PTEN. pRS315 and pRS316 are CEN/ARS LEU2 and URA3 vectors, respectively, and they were used as control vectors.13
Yeast strains and media
Yeast strains used were YPH499 (MATa, ura3‐52, lys2‐801, ade2‐101, trp1‐Δ63, his3‐Δ200, leu2‐Δ1) and AD1‐9 (MATα, Δyor1, Δsnq2, Δpdr5, Δpdr10, Δpdr11, Δycf1, Δpdr3, Δpdr15, Δpdr1, Δhis1, Δura3). YPH499 was purchased from Stratagene (La Jolla, CA, USA). AD1‐9 was gifted by Nader Nourizad (Stanford University, Stanford, CA, USA). In AD1‐9, to append another auxotrophic selection marker, generating AD1‐9_leu2, LEU2 was disrupted by means of a hisG‐URA3‐hisG cassette.14 A general non‐selective yeast growth medium, YPD broth, or agar was used. Synthetic minimal medium (SD) contained glucose and lacked auxotrophic markers uracil (U) and leucine (L). In synthetic galactose medium (SGal), glucose was replaced with galactose.
Chemical library
The SCADS deposited chemical library was kindly provided by SCADS, supported by a Grant‐in‐Aid for Scientific Research on Innovative Areas and Scientific Support Programs for Cancer Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The library was composed of three 96‐well plates. All compounds were dissolved in DMSO.
Chemicals
LY294002 and suberoylanilide hydroxamic acid (SAHA) were purchased from Cayman Chemical (Ann‐Arbor, MI, USA). Trichostatin A was purchased from Sigma‐Aldrich (St. Louis, MI, USA). FK228 and its analogs were synthesized and provided by T. K.
Screening system for PI3K inhibitors using S. cerevisiae
The sets of plasmids, pRS316 and pRS315, pSJ01 (expressing p110α) and pRS315, and pSJ01 and pSJ21 (coexpressing p110α and PTEN) were transformed into AD1‐9_leu2. The transformants were incubated overnight in SD‐U‐L and adjusted to an A 600 of 0.3. We inoculated 10 μL transformant cells to 200 μL aliquots of SGal‐U‐L media containing each test compound. The aliquots were then incubated with shaking in 96‐well plates for 24 h at 30°C. Compounds that showed an increase in A 600 value among the transformants expressing p110α were identified as candidates.
In vitro PI3K assay
PI3K activity was evaluated by an electrophoretic mobility shift assay (Carna Biosciences, Kobe, Japan). Compound solutions (1000 nM PIP2, 50 μM ATP, 5 mM MgCl, and 21 nM PI3K [p110α/p85α]) were prepared with assay buffer containing 2 mM DTT (Carna Biosciences) and mixed and incubated in a 384‐well microplate for 5 h at room temperature. To evaluate the ATP‐competitive effect, PI3K activity was also examined under a higher concentration of ATP (500 μM) and a shorter incubation period (2 h). Reaction mixtures were then applied to the LabChip (Caliper Life Sciences, Hopkinton, MA, USA), and the product, PIP3, and substrate, PIP2, peaks were separated and quantified. The PI3K reaction was evaluated by the product ratio, calculated from peak heights of the product and substrate.
Molecular modeling of the PI3K–FK228 complex
Construction and conformational analysis of the PI3K–FK228 complex structure models were carried out using eHiTS (SimBioSys, San Francisco, CA, USA). The structure of the PI3K (p110α H1047R/p85α)–wortmannin complex (Protein Data Bank Identification Code: 3HHM) was used to obtain the PI3K template structure. FK228 is a pro‐drug and is structurally changed in cells to a reduced active form for HDAC inhibition.15 Therefore, the structure of reduced FK228 was applied in this docking simulation. The top‐ranked docking model, according to the eHiTS score, was adopted and displayed.
Cell lines
The human cancer cell lines used in this study were the prostate cancer cell line PC3 (Cell Resource Center, Institute of Development, Aging, and Cancer, Tohoku University, Sendai, Japan) and the colorectal cancer cell lines HCT116 and RKO (both ATCC, Manassas, VA, USA), and CO115 (kindly provided by John M. Mariadason, Ludwig Institute for Cancer Research, Melbourne, Vic., Australia). Cells were cultured in RPMI‐1640 medium (Sigma‐Aldrich) containing 10% FBS.
Western blot analysis
Cells treated with test compounds were harvested and lysed in a buffer (500 mM Tris‐HCl [pH 7.5], 100 mM NaCl, 2 mM EDTA, 1 mM sodium orthovanadate, 1% Tergitol‐type NP‐40, and 1% protease inhibitor cocktail; Sigma‐Aldrich). Cell lysates were separated by SDS‐PAGE, followed by electroblotting onto PVDF membranes (Millipore, Billerica, MA, USA). Rabbit polyclonal antibodies for AKT, phosphorylated AKT (Ser473), phosphorylated AKT (Thr308), phosphorylated GSK‐3β (Ser9), phosphorylated mTOR (Ser2448), phosphorylated p70S6K (Thr389), phosphorylated 4E‐BP1 (Thr37/46), phosphorylatedMEK1/2 (Ser217/221), and phosphorylated ERK1/2 (Thr202/Tyr204) were all purchased from Cell Signaling Technology (Danvers, MA, USA). Other antibodies were purchased as follows: anti‐β‐actin monoclonal antibody (Sigma‐Aldrich), anti‐PARP1/2 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐acetyl‐Histone H3 and anti‐acetyl‐histone H4 (Upstate Biotechnology, Lake Placid, NY, USA), and Alexa Fluor 680 as a secondary antibody (Invitrogen, Carlsbad, CA, USA). Immunoreactive bands were detected using the Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA).
Cell proliferation assay
The colorectal cancer cell lines HCT116, CO115, and RKO were used. A total of 8 × 103 cells per well were seeded and incubated into 96‐well plates for 24 h. They were then treated with compounds and further cultured for 24 h at 37°C. Cell viability was assayed by quantifying the uptake and digestion of 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium monosodium salt (Dojindo Laboratories, Kumamoto, Japan) using a SpectraMax M2e. We calculated the ratio of surviving cells to controls treated with 1% DMSO alone.
Cell cycle analysis by FACS
HCT116 cells were seeded into six‐well plates at a concentration of 2 × 105 per well, exposed to the compounds at various concentrations, cultured for 24 h, collected, and sorted by FACS using the Cytomics FC 500 flow cytometry system (Beckman Coulter, Brea, CA, USA).
Results
Human PI3K induces growth inhibition in S. cerevisiae
Ectopic expression of human p110α inhibited the growth of the YPH499 strain of yeast. In contrast, coexpression of wild‐type PTEN in p110α‐expressing yeast rescued this growth inhibition (Fig. 1A,B). In YPH499, the PI3K inhibitor LY294002 partially rescued the growth inhibition in the transformants expressing p110α (Fig. 1B), as previously reported.10 Similar results were observed with AD1‐9 (data not shown), a drug‐sensitive strain due to the deletion of seven ABC transporters.16 We compared the sensitivity of AD1‐9 to the PI3K inhibitor LY294002 with that of the wild‐type strain YPH499. The growth inhibition induced by p110α expression was rescued at a LY294002 concentration of 50 μM in YPH499 (Fig. 1C); in contrast, this rescue was achieved at 5 μM LY294002 in AD1‐9 (Fig. 1D). This result indicates that AD1‐9 is approximately 10 times more sensitive to LY294002 compared to YPH499; therefore, AD1‐9 was used for drug screening.
Figure 1.

Identification of FK228 and its analogs as candidate phosphatidylinositol 3‐kinase (PI3K) inhibitors using Saccharomyces cerevisiae. (A) YPH499 was cotransformed with the following sets of URA3 and LEU2 marked vectors: top row, pRS316 and pRS315; second row, pSJ01 (p110α) and pRS315; and third row, pSJ01 and pSJ21 (PTEN). Serial 10‐fold dilutions of the transformants were deposited on solid synthetic minimal medium–uracil–leucine (SD‐U‐L) and synthetic galactose medium (SGal)‐U‐L media. (B) YPH499 transformants were incubated in SGal‐U‐L medium, with or without 50 μM LY294002. Then A 600 was measured as an indicator of yeast growth. (C,D) Transformants of YPH499 and AD1‐9 expressing p110α were cultured in SGal‐U‐L medium containing various concentrations of LY294002. Sensitivity for LY294002 was compared by detecting the concentration at which growth inhibition was rescued (arrow). (E) Screening results of the chemical library of the Screening Committee of Anticancer Drugs. AD1‐9 transformants expressing p110α were cultured in SGal‐U‐L media containing each compound. A 600 values are the mean of experiments carried out in triplicate. Test compounds are arranged according to A 600 values in the figure. LY294002, DMSO, and FK228 and its analogs are indicated.
Screening of chemical library
We screened the SCADS deposited chemical library using the yeast screening system. All compounds were tested at two different concentrations, 0.5 and 5 μM. No compounds recovered the cells at a concentration of 0.5 μM (data not shown). However, at a concentration of 5 μM, some compounds rescued the p110α‐induced growth inhibition, as with LY294002 (Fig. 1E). As a result, we identified seven compounds as candidate PI3K inhibitors; these included an HDAC inhibitor, FK228, and its analogs (Fig. 1E). The structures of FK228 and its analogs and their IC50 values for HDAC1 and HDAC6 examined by SCADS are shown in Figure 2. All of them strongly inhibited HDAC1 in a selective manner.
Figure 2.
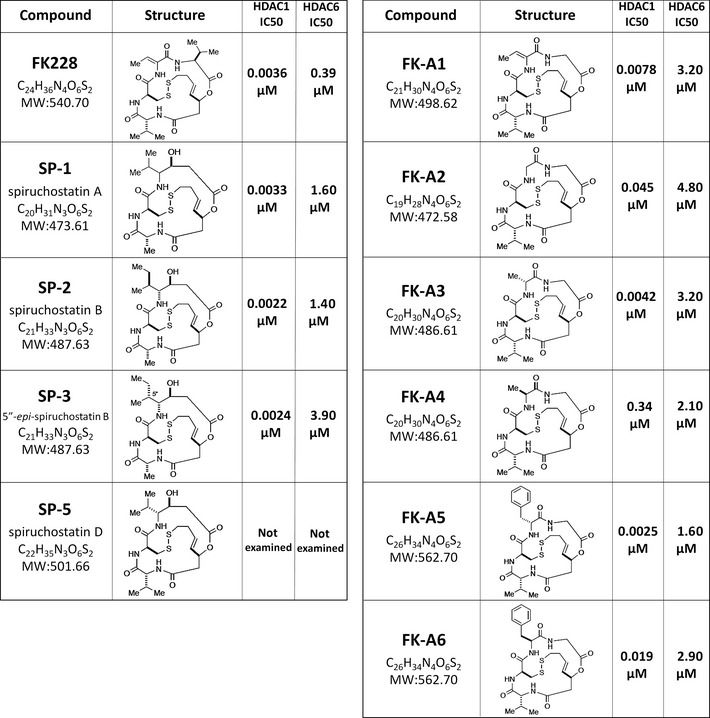
Structures and histone deacetylase (HDAC) inhibitory activity of FK228 and its analogs. The structures of FK228 and the analogs examined in this study and their IC 50 values for HDAC1 and HDAC6 are shown.
Inhibition of PI3K activity by FK228 and its analogs
Eleven compounds including the seven screened compounds (FK228, SP‐1, SP‐2, SP‐3, FK‐A1, FK‐A3, and FK‐A5) and four other FK228 analogs (SP‐5, FK‐A2, FK‐A4, and FK‐A6) were examined for PI3K inhibition at a concentration of 20 μM by mobility shift assay. All compounds directly inhibited PI3K activity (Fig. 3A). Of these, FK‐A5 was the most potent, whereas SP‐3 was the weakest (Fig. 3A). Next, we determined the IC50 for PI3K activity. The IC50 values of FK228, FK‐A5, SP‐3, and LY294002 were 57.1, 26.2, 107.0 and 0.7 μM, respectively (Fig. 3B–E). FK‐A5 showed double the activity of FK228 and 4‐fold the activity of SP‐3, but its activity was 37‐fold less than that of LY294002. These data indicate that FK228 and its analogs directly inhibit PI3K activity, but that the potency of inhibitory activity differs among analogs.
Figure 3.
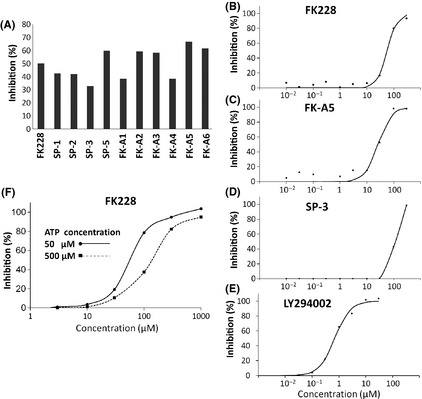
Effects of FK228 and its analogs on phosphatidylinositol 3‐kinase (PI3K) activity. (A) Inhibition of PI3K activity was evaluated by mobility shift assay. The read‐out value of a control reaction (complete reaction mixture) was set as a 0% inhibition, and the percentage inhibition of each test solution was calculated at a concentration of 20 μM. Data are the mean of two experiments carried out in duplicate. (B–E) IC 50 values of FK228, FK‐A5, SP‐3, and LY294002 were calculated from concentration versus % inhibition curves by fitting to a four‐parameter logistic curve. (F) Inhibition of PI3K activity of FK228 was evaluated under the conditions of two different ATP concentrations, 50 and 500 μM. Each concentration versus % inhibition curve is shown.
Molecular modeling of the PI3K–FK228 complex
All known PI3K inhibitors are ATP‐competitive inhibitors;17 therefore, we investigated whether FK228 was capable of binding to the ATP‐binding site of p110α. As shown in Figure 4, the ATP‐binding pocket of p110α was occupied by reduced FK228. Additionally, PI3K inhibitory activity of FK228 was weakened under the high concentration of ATP in vitro assay (Fig. 3F). The IC50 values of FK228 under the standard ATP concentration (50 μM) and high ATP concentration (500 μM) were 57.2 and 125.0 μM, respectively. These data suggest that FK228 is an ATP‐competitive PI3K inhibitor.
Figure 4.
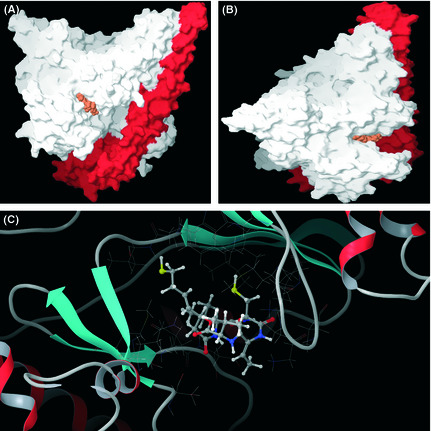
Molecular modeling of the phosphatidylinositol 3‐kinase (PI3K)–FK228 complex. (A) Molecular surface structure of the PI3K–FK228 complex model. Orange, reduced form of FK228; red, p85α; white, p110α. (B) Side view of Fig. 4(A). (C) Enlarged picture around the ligand. A ball and stick model shows the reduced FK228 in the PI3K ribbon and line drawing.
Inhibition of AKT phosphorylation and downstream signaling by FK228 and FK‐A5
We used PC3 cells to evaluate FK228 and FK‐A5‐mediated inhibition of the AKT pathway by Western blotting, because PC3 cells are PTEN‐null and show constitutively active AKT.18 FK228 and FK‐A5 as well as LY294002 inhibited the AKT phosphorylation within 30 min, without altering total AKT levels, at a concentration of 10 μM (Fig. 5A). Next, we investigated the effect of various concentrations of FK228 and FK‐A5 on AKT and its downstream signaling components. FK228 and FK‐A5 inhibited phosphorylation of AKT, GSK‐3β, mTOR, p70S6K, and 4E‐BP1 in a dose‐dependent manner at μM‐range concentrations. As for the Ras‐MAP kinase pathway, another major signaling pathway involved in cell proliferation, the level of phosphorylated MEK1/2 was unaltered, but the level of phosphorylated ERK1/2 was reduced slightly by the addition of 30 μM FK228 or FK‐A5. These results indicate that FK228 and FK‐A5 act primarily through the AKT signaling pathway and that they are capable of exerting inhibitory activity on other kinases.
Figure 5.
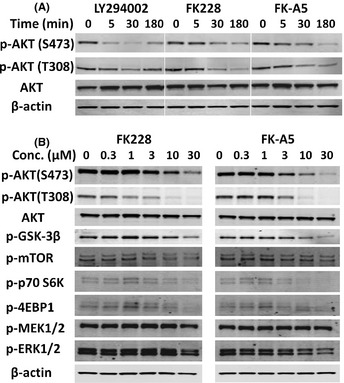
Western blot analysis of protein kinase B (AKT) and its downstream components in cells treated with FK228 or FK‐A5. (A) Evaluation of the experimental time course. PC3 cells were treated with LY294002, FK228, or FK‐A5 at a concentration of 10 μM. (B) Effect of various concentrations of FK228 or FK‐A5 on AKT and its downstream signaling components. PC3 cells were treated with FK228 or FK‐A5 for 3 h.
Antiproliferative effects of FK228, FK‐A5, and SP‐3
Among the FK228 analogs, FK‐A5 is the most potent PI3K inhibitor, and SP‐3 is the weakest (Fig. 3A). However, those analogs retain almost equal activity as HDAC inhibitors (Fig. 2). To examine the antiproliferative effects of FK228, FK‐A5, and SP‐3, we used the HDAC inhibitor‐resistant cell lines RKO and CO115 as well as the HDAC inhibitor‐sensitive cell line HCT116.19, 20 HCT116, RKO, and CO115 are colorectal cancer cells with microsatellite instability. In RKO and CO115 cells, biallelic frameshift mutations occur in the (A)9 repeat of exon 1 of HDAC2, leading to the loss of HDAC2.19, 20 This loss induces apoptotic protease‐activating factor‐1 expression, which dysregulates apoptosis and renders the cells resistant to HDAC inhibitors.20 In HDAC inhibitor‐sensitive HCT116 cells, SAHA at a concentration of 2.5 μM, and FK228, FK‐A5, and SP‐3 at nM‐range concentrations inhibited cell growth by 30–60% (Fig. 6). In contrast, CO115 and RKO were more resistant to these HDAC inhibitors than HCT116 cells (Fig. 6). Similar results were observed when 250 nM trichostatin A was used as another HDAC inhibitor (Fig. S1). All three cell lines examined were sensitive to LY294002, which reduced cell counts by 41–51%. The combination of LY294002 and SAHA caused enhanced antiproliferative effects in HCT116 cells, but not in RKO or CO115 cells. The combination of LY294002 and FK228, FK‐A5, or SP‐3 at nM‐range concentrations resulted in enhanced antiproliferative effects in HCT116 cells. Conversely, in RKO and CO115 cells, these combinations did not enhance antiproliferative effects, resulting in an effect almost identical to that of LY294002 monotherapy. At concentrations of 5 and 50 μM to inhibit PI3K activity, FK228 or FK‐A5 monotherapy showed potent antiproliferative (cytotoxic) effects, reducing 80–90% of cell counts, even in HDAC inhibitor‐resistant cells. However, SP‐3, with its poor PI3K inhibitory activity, did not potentiate the antiproliferative effect at μM‐range concentrations. These results suggest that inhibition of PI3K activity enhances the cytotoxicity of FK228 or FK‐A5 at μM‐range concentrations.
Figure 6.
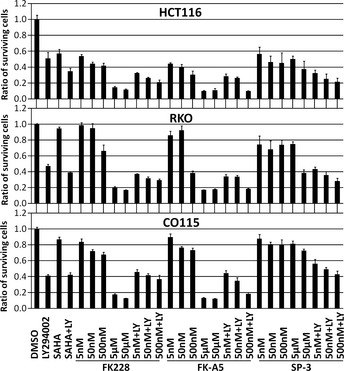
Antiproliferative effects of FK228, FK‐A5, and SP‐3. HCT116, RKO, and CO115 colorectal cancer cells were treated with: DMSO; 50 μM LY294002 (LY); 2.5 μM suberoylanilide hydroxamic acid (SAHA); a combination of 50 μM LY with 2.5 μM SAHA; FK228, FK‐A5, or SP‐3 at concentrations of 5, 50, 500 nM, 5, and 50 μM; and a combination of 50 μM LY with FK228, FK‐A5, or SP‐3 at concentrations of 5, 50, and 500 nM. The cell viability was assayed 24 h after treatment. The ratio of surviving cells to the controls treated with 1% DMSO was calculated.
Effects of FK228, FK‐A5, and SP‐3 on apoptosis and cell cycle
We carried out a FACS analysis of cell death induced by FK228 and FK‐A5. HCT116 cells were treated with LY294002, SAHA, FK228, or FK‐A5 under the same conditions as in the cell proliferation assay. The cell cycle phases at which HDAC inhibitors induce arrest are reported to differ depending on the drug concentration and cell line.21, 22, 23 In this experiment, SAHA induced G2/M arrest in HCT116 cells (Fig. 7A). FK228 and FK‐A5 at nM‐range concentrations also induced G2/M arrest (Fig. 7B). In contrast, inhibition of PI3K was reported to induce G0/G1 arrest.24, 25 In agreement with the previous reports, LY294002 induced G0/G1 arrest (Fig. 7A). The combination of LY294002 and SAHA, and combinations of LY294002 with either FK228 or FK‐A5 at nM‐range concentrations increased the sub‐G1 fraction to approximately 40%, indicating apoptosis induction (Fig. 7A,B). At a concentration of 5 μM, FK228 or FK‐A5 increased the sub‐G1 fraction to 37%, and the DNA histograms were similar to those of combination therapy with LY294002 and an HDAC inhibitor (Fig. 7A,B). Apoptosis induction by FK228 and FK‐A5 was supported by increased expression of cleaved PARP (Fig. 7C). FK228 and FK‐A5 at a concentration of 5 μM caused the greatest elevation in the level of cleaved PARP, potently inducing apoptosis, in accordance with the results from the FACS analysis. Furthermore, FK228 and FK‐A5 at this concentration simultaneously inhibited AKT phosphorylation and accelerated histone acetylation (Fig. 7C). Similar results were observed in CO115 treated with FK‐A5 (Fig. 7D). In contrast, SP‐3 did not inhibit AKT phosphorylation, although it acetylated histone (Fig. 7D). These results indicate that μM‐range concentrations of FK228 and FK‐A5 potently induce apoptosis through HDAC/PI3K dual inhibition.
Figure 7.
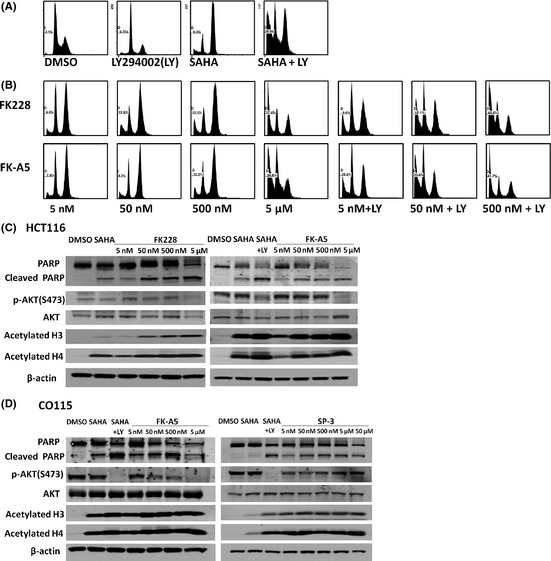
Effects of FK228, FK‐A5, and SP‐3 on apoptosis, cell cycle, protein kinase B (AKT) phosphorylation, and histone acetylation. (A,B) HCT116 colorectal cancer cells were treated with: DMSO; 50 μM LY294002 (LY); 2.5 μM suberoylanilide hydroxamic acid (SAHA); a combination of 50 μM LY with 2.5 μM SAHA; FK228 or FK‐A5 at concentrations of 5, 50, 500 nM, and 5 μM; and a combination of 50 μM LY with FK228 or FK‐A5 at concentrations of 5, 50, and 500 nM. The cells were harvested 24 h after treatment. (C,D) Western blot analysis of poly(ADP‐ribose) polymerase (PARP) cleavage, AKT dephosphorylation, and histone acetylation by FK228, FK‐A5, or SP‐3 in HCT116 and CO115 cells cultured as indicated in Fig. 6.
Discussion
In this study, we identified FK228 and its analogs as novel PI3K inhibitors. FK228 is a potent HDAC inhibitor, with recent approval for use in the treatment of cutaneous T‐cell lymphoma.11 The HDAC inhibitors have pleiotropic antitumor activities because of their ability to act on non‐histone targets.12 However, until now, there have been no reports documenting its function as a kinase inhibitor. In this study, we add a new mechanism of direct PI3K inhibition to the pleiotropic functions of FK228 in antitumor activity.
Kodani et al.26 reported that 2–10 nM FK228 inhibited AKT phosphorylation and suppressed the PI3K/AKT pathway in A549 lung cancer cells, although they did not elucidate the mechanism for this in their study. They showed that 5 nM FK228 dephosphorylated AKT in 24 h, but not in 6 or 12 h.26 In contrast, we showed that AKT dephosphorylation by FK228 began in 5 min, in a manner similar to that for LY294002 (Fig. 5A). In addition, we showed that the concentration at which FK228 had PI3K inhibitory activity was within the μM‐range. Thus, we speculated that the suppression of the AKT pathway by FK228 reported by Kodani et al.26 does not occur by direct inhibition of PI3K, but through another indirect mechanism, such as epigenetic alteration. This study is the first to show that FK228 directly inhibits PI3K activity.
FK228 is structurally changed in cells to a reduced active form for HDAC inhibition.15 Therefore, it is assumed that the reduced FK228 is also active for PI3K inhibition. Supporting that, in vitro PI3K assay was carried out under conditions containing 2 mM DTT which is expected to reduce FK228 and its analogs. In Figures 5 and 7, FK228 and FK‐A5 inhibited the AKT signaling pathway in cells at lower concentrations than in vitro IC50 values. We speculated that reduction power in vitro might be insufficient compared to that in cells, for changing FK228 and FK‐A5 to active form.
The results of the cell proliferation assay revealed that μM‐range concentrations of FK228 or FK‐A5 resulted in potent cytotoxicity, even in HDAC inhibitor‐resistant cells (Fig. 6). Furthermore, the results of FACS and Western blotting showed that the cause of cell death was apoptosis. We suggest that HDAC/PI3K dual inhibition by FK228 or FK‐A5 at μM‐range concentrations contributes to the potent apoptosis induction because: (i) FK228 and FK‐A5 at μM concentrations inhibited AKT phosphorylation and acetylated histones simultaneously (Fig. 7C,D); (ii) both apoptosis and the effects on the cell cycle induced by FK228 or FK‐A5 at μM‐range concentrations mimicked the effects of a combination of individual HDAC and PI3K inhibitors as measured by FACS analyses (Fig. 7A,B); and (iii) SP‐3 with poor PI3K inhibitory activity could not potentiate the cytotoxic effect. The combination of an HDAC inhibitor with a PI3K inhibitor was shown to result in cytotoxic synergy27, therefore, a HDAC/PI3K dual inhibitor could contribute greatly to the treatment of refractory cancer.
FK228 and its analogs are potent HDAC inhibitors, capable of inhibiting HDAC1 at nM‐range concentrations.28 In contrast, the PI3K inhibitory activity of these compounds is shown within the μM range. Of these analogs, FK‐A5 and FK‐A6 were the most potent PI3K inhibitors (Fig. 3A). FK‐A5 and FK‐A6 are stereoisomers and have a characteristic structure, possessing a benzyl group in position 7 (Fig. 2). If their structural differences are found to be responsible for the greater potency compared with other analogs, this would provide us with a clue regarding optimization of the PI3K inhibitory activity of these compounds.
In conclusion, we showed that FK228 and its analogs directly inhibit PI3K activity and potently induce apoptosis through HDAC/PI3K dual inhibition at μM‐range concentrations. Further studies are required to identify analogs that can inhibit PI3K more potently.
Disclosure Statement
Chikashi Ishioka received a research grant from Chugai Pharmaceutical Co. and Novartis Pharma K.K., and also recieved lecture fees from Taiho Pharmaceutical Co. None of the other authors has any conflict of interest.
Supporting information
Fig. S1. Antiproliferative effects of trichostatin A.
{kind=link}
Acknowledgments
This study was supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan, the Ministry of Health, Labour and Welfare, Japan, and the Japan Science and Technology Agency.
(Cancer Sci 2012; 103: 1994–2001)
References
- 1. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem 1998; 67: 481–507. [DOI] [PubMed] [Google Scholar]
- 2. Cantley LC. The phosphoinositide 3‐kinase pathway. Science 2002; 296: 1655–7. [DOI] [PubMed] [Google Scholar]
- 3. Samuels Y, Wang Z, Bardelli A et al High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. [DOI] [PubMed] [Google Scholar]
- 4. Ikenoue T, Kanai F, Hikiba Y et al Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 2005; 65: 4562–7. [DOI] [PubMed] [Google Scholar]
- 5. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5‐trisphosphate. J Biol Chem 1998; 273: 13375–8. [DOI] [PubMed] [Google Scholar]
- 6. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell 2008; 133: 403–14. [DOI] [PubMed] [Google Scholar]
- 7. Kong D, Yamori T. Advances in development of phosphatidylinositol 3‐kinase inhibitors. Curr Med Chem 2009; 16: 2839–54. [DOI] [PubMed] [Google Scholar]
- 8. Barberis A, Gunde T, Berset C, Audetat S, Luthi U. Yeast as a screening tool. Drug Discovery Today: Technologies 2005; 2: 187–92. [DOI] [PubMed] [Google Scholar]
- 9. Cid V, Rodríguez‐Escudero I, Andrés‐Pons A et al Assessment of PTEN tumor suppressor activity in nonmammalian models: the year of the yeast. Oncogene 2008; 27: 5431–42. [DOI] [PubMed] [Google Scholar]
- 10. Rodríguez‐Escudero I, Roelants FM, Thorner J, Nombela C, Molina M, Cid VJ. Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast. Biochem J 2005; 390: 613–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bertino EM, Otterson GA. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin Investig Drugs 2011; 20: 1151–8. [DOI] [PubMed] [Google Scholar]
- 12. Xu W, Parmigiani R, Marks P. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26: 5541–52. [DOI] [PubMed] [Google Scholar]
- 13. Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics 1989; 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of USR3 selection in the construction of multiply disrupted yeast strains. Genetics 1987; 116: 541–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Furumai R, Matsuyama A, Kobashi N et al FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res 2002; 62: 4916–21. [PubMed] [Google Scholar]
- 16. Rogers B, Decottignies A, Kolaczkowski M, Carvajal E, Balzi E, Goffeau A. The pleitropic drug ABC transporters from Saccharomyces cerevisiae . J Mol Microbiol Biotechnol 2001; 3: 207–14. [PubMed] [Google Scholar]
- 17. Walker EH, Pacold ME, Perisic O et al Structural determinants of phosphoinositide 3‐kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 2000; 6: 909–19. [DOI] [PubMed] [Google Scholar]
- 18. Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res 2002; 62: 6141–5. [PubMed] [Google Scholar]
- 19. Ropero S, Fraga MF, Ballestar E et al A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet 2006; 38: 566–9. [DOI] [PubMed] [Google Scholar]
- 20. Hanigan CL, van Engeland M, De Bruine AP et al An inactivating mutation in HDAC2 leads to dysregulation of apoptosis mediated by APAF1. Gastroenterology 2008; 135: 1654–64. [DOI] [PubMed] [Google Scholar]
- 21. Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene‐associated histone acetylation. Proc Natl Acad Sci U S A 2000; 97: 10014–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res 1998; 241: 126–33. [DOI] [PubMed] [Google Scholar]
- 23. Hartlapp I, Pallasch C, Weibert G, Kemkers A, Hummel M, Re D. Depsipeptide induces cell death in Hodgkin lymphoma‐derived cell lines. Leuk Res 2009; 33: 929–36. [DOI] [PubMed] [Google Scholar]
- 24. Dan S, Yoshimi H, Okamura M, Mukai Y, Yamori T. Inhibition of PI3K by ZSTK474 suppressed tumor growth not via apoptosis but G0/G1 arrest. Biochem Biophys Res Commun 2009; 379: 104–9. [DOI] [PubMed] [Google Scholar]
- 25. Reagan‐Shaw S, Ahmad N. RNA interference‐mediated depletion of phosphoinositide 3‐kinase activates forkhead box class O transcription factors and induces cell cycle arrest and apoptosis in breast carcinoma cells. Cancer Res 2006; 66: 1062–9. [DOI] [PubMed] [Google Scholar]
- 26. Kodani M, Igishi T, Matsumoto S et al Suppression of phosphatidylinositol 3‐kinase/Akt signaling pathway is a determinant of the sensitivity to a novel histone deacetylase inhibitor, FK228, in lung adenocarcinoma cells. Oncol Rep 2005; 13: 477–83. [PubMed] [Google Scholar]
- 27. Wozniak MB, Villuendas R, Bischoff JR et al Vorinostat interferes with the signaling transduction pathway of T‐cell receptor and synergizes with phosphoinositide‐3 kinase inhibitors in cutaneous T‐cell lymphoma. Haematologica 2010; 95: 613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Narita K, Kikuchi T, Watanabe K et al Total synthesis of the bicyclic depsipeptide HDAC inhibitors Spiruchostatins A and B, 5”‐epi‐Spiruchostatin B, FK228 (FR901228) and preliminary evaluation of their biological activity. Chem Eur J 2009; 15: 11174–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Antiproliferative effects of trichostatin A.