Abstract
Free full text
An alpha 2 collagen VIII transgenic knock-in mouse model of Fuchs endothelial corneal dystrophy shows early endothelial cell unfolded protein response and apoptosis
Abstract
Fuchs endothelial corneal dystrophy (FECD) is a leading indication for corneal transplantation. FECD is characterized by progressive alterations in endothelial cell morphology, excrescences (guttae) and thickening of the endothelial basement membrane and cell death. Ultimately, these changes lead to corneal edema and vision loss. Due to the lack of vision loss in early disease stages and the decades long disease course, early pathophysiology in FECD is virtually unknown as studies of pathologic tissues have been limited to end-stage tissues obtained at transplant. The first genetic defect shown to cause FECD was a point mutation causing a glutamine to lysine substitution at amino acid position 455 (Q455K) in the alpha 2 collagen 8 gene (COL8A2) which results in an early onset form of the disease. Homozygous mutant knock-in mice with this mutation (Col8a2Q455K/Q455K) show features strikingly similar to human disease, including progressive alterations in endothelial cell morphology, cell loss and basement membrane guttae. Ultrastructural analysis shows the predominant defect as dilated endoplasmic reticulum (ER), suggesting ER stress and unfolded protein response (UPR) activation. Immunohistochemistry, western blotting, quantitative reverse transcriptase polymerase chain reaction and terminal deoxynucleotidyl transferase 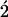 -deoxyuridine,
-deoxyuridine,  -triphosphate nick end-labeling analyses support UPR activation and UPR-associated apoptosis in the Col8a2Q455K/Q455K mutant corneal endothelium. This study confirms the Q455K substitution in the COL8A2 gene as being sufficient to cause FECD in the first mouse model of this disease and supports the role of the UPR and UPR-associated apoptosis in the pathogenesis of FECD caused by COL8A2 mutations.
-triphosphate nick end-labeling analyses support UPR activation and UPR-associated apoptosis in the Col8a2Q455K/Q455K mutant corneal endothelium. This study confirms the Q455K substitution in the COL8A2 gene as being sufficient to cause FECD in the first mouse model of this disease and supports the role of the UPR and UPR-associated apoptosis in the pathogenesis of FECD caused by COL8A2 mutations.
INTRODUCTION
Fuchs endothelial corneal dystrophy (FECD) is a degenerative disease of the corneal endothelium which affects an estimated 5% of the US population and is one of the most common indications for corneal transplantation (1). FECD is characterized by corneal endothelial cell (CEC) loss and abnormalities, including drop-like excrescences (guttae) and thickening of the endothelial basement membrane (Descemet membrane, DM). With advanced CEC loss, corneal edema, clouding and vision loss occur, and corneal transplantation is the only effective treatment (2). Due to the decades-long disease course, absence of symptoms in milder stages and lack of a disease model, studies of early pathologic tissue have been infeasible. Instead, studies of FECD pathophysiology have used end-stage corneas obtained at transplantation. Better understanding of the early pathophysiology of FECD through an accurate in vivo model should facilitate the development of treatment options to delay or prevent the need for corneal transplantation surgery with its attendant risks, complications and costs.
The genetic basis of FECD has long been recognized (3,4).The first mutations shown to cause FECD were associated with an early onset form of the disease and were identified in the alpha 2 collagen VIII (COL8A2) gene. These mutations included a cytosine to adenine missense mutation specifying a glutamine to lysine change at amino acid 455 (p.Q455K) (5). Additional COL8A2 mutations associated with FECD include a glutamine to valine change at amino acid 455 and a leucine to tryptophan change at amino acid 450 (6,7). Altogether, COL8A2 FECD mutations are rare and have been described in a total of 11 pedigrees and 6 sporadic cases (5–8). FECD mutations have also been identified in the SLC4A11, TCF4 and TCF8 genes (1,9,10) (for a complete listing of FECD mutations and chromosome loci, see www.ncbi.nlm.nih.gov/omim/136800).
Previously, we described activation of the unfolded protein response (UPR) (11,12) in genetically undifferentiated, end-stage FECD corneas (13). Protein folding is critical for cellular functioning, and eukaryotic cells have conserved mechanisms within the endoplasmic reticulum (ER) to ensure proper protein folding and disposal of cytotoxic misfolded proteins (11). The UPR is a comprehensive program to reduce the accumulation of toxic unfolded proteins (12,14). Mediators of the UPR include glucose response protein 78 (GRP78), activating transcription factor 4 (ATF4) and growth arrest and DNA damage 153 (GADD153), also known as C/EBP homologous protein (12,14). Although the initial function of the UPR is to restore a physiologic level of correctly folded proteins within the cell, severe and unresolved unfolded protein stress can initiate an apoptotic function of the UPR. UPR-associated apoptosis likely occurs via multiple pathways, one of which includes caspase 12, an ER-associated proximal effector of the caspase cascade which subsequently activates caspases 9 and 3, leading to apoptosis (15–17).
Based on previous work supporting the mouse as a model for human corneal endothelial diseases (18), we produced a knock-in mouse containing a point mutation homologous to the human Q455K COL8A2 mutation in order to assess whether the UPR is activated in early FECD corneal endothelium. Our results demonstrate endothelial cell UPR activation and UPR-associated apoptosis in the first in vivo model of FECD, further supporting a central role for the UPR in the pathogenesis of FECD at an earlier disease stage than has been studied previously. Furthermore, our findings establish that the p.Q455K substitution in COL8A2 is both necessary and sufficient to cause early onset FECD in humans and further validate the mouse as a model system for human corneal endothelial diseases.
RESULTS
Generation of Col8a2 Q455K knock-in mice
The mouse Col8a2 protein (NCBI reference sequence NP_955767.1) is a 698-amino acid protein which demonstrates 94% amino acid identity (protein BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi) to the 702-amino acid human COL8A2 protein (NCBI reference sequence NP_005193.1). A homologous glutamine residue at amino acid position 455 in the human COL8A2 protein corresponds to amino acid 451 in the mouse protein. Alignment of mouse and human proteins shows identical residues 47 amino acids to the amino terminus and 40 amino acids to the carboxyl terminus of the homologous human 455/mouse 451 amino acid positions.
A targeting vector containing the Col8a2 Q455K mutation was generated using a recombineering-based approach (19) and site-directed mutagenesis (Fig. 1A, see Materials and Methods). F1 heterozygous mutants before and after excision of the neomycin-resistance selection cassette (Materials and Methods) were confirmed by Southern analysis (Fig. 1B). Direct sequence analysis and diagnostic restriction enzyme digestion confirmed the cytosine to adenine base change corresponding to the p.Q455K substitution (Fig. 1C and D). Homozygous mutant mice appear to have normal overall phenotype, development and breeding. No extra-corneal abnormalities were observed.

Generation of Col8a2 Q455K knock-in mice. (A) Map of targeting strategy. The targeting vector containing a portion of Col8a2 intron 2, the entire exon 3 (Ex3) and intron 3 and a portion of exon 4 on mouse chromosome (chr) 4 was produced as described in the Materials and Methods. The targeting vector contained the Col8a2 Q455K mutation (asterisk) in exon 4 and an additional diagnostic MfeI site in intron 3. Chimeric animals were bred to heterozygosity and subsequently crossed with a cre-recombinase expressing line. (B) Southern analysis of MfeI-digested genomic DNA from two different (i) F1 WT, (ii) F1 Col8a2WT/Q455K heterozygous and (iii) F2 Col8a2WT/Q455K heterozygous animals. F1 WT mice show an 18.1 kb band (i) and F1 heterozygotes show an additional 6.1 kb band for the mutant allele (ii). F2 heterozygotes show cre-mediated excision of the pGK-neomycin cassette and a smaller 4.2 kb mutant band (iii). (C) Sequence analysis of Col8a2WT/Q455K mutant mice. Both the WT and mutant sequences (C/A) are seen with equal chromatogram signal intensity (arrow). (D) Fnu4HI restriction enzyme digestion of PCR fragments showing two different Col8a2Q455K/Q455K (MUT, 1–2) and WT (3–4) mice. The Q455K mutation results in a 574bp band, and the WT sequence results in a 384 bp band.
Morphologic characterization of Col8a2 Q455K/Q455K corneal endothelium
Clinical confocal microscopy of 5- and 10-month wild-type (WT) mouse endothelium (Fig. 2A and C) shows a characteristic polygonal monolayer with regular size and shape comparable with normal human endothelium (Fig. 2E). In contrast, 5- and 10-month Col8a2Q455K/Q455K (MUT) endothelium (Fig. 2B and D) shows progressive variability in size and shape, accumulation of DM excrescences (guttae) highly characteristic of FECD and a highly similar appearance to endothelium from a human patient heterozygous for the COL8A2 Q455K mutation (Fig. 2F). Central CEC densities (Fig. 2G), a clinical measure of endothelial health, showed a 27% reduction (1552 ± 241 versus 2137 ± 155 cells/mm2, mean ± standard deviation) in 5-month and a 41% reduction (1240 ± 265 versus 2101 ± 120 cells/mm2) in 10-month MUT versus WT mice. CEC density showed a 20% reduction in 10- versus 5-month MUT mice (Fig. 2G). Percent hexagonal cells (Fig. 2H), a measure of normal cellular morphology, showed a 31% reduction (54 ± 14 versus 78 ± 10%) in 5-month and a 57% reduction (31 ± 20 versus 72 ± 6%) in 10-month MUT versus WT mice.

Characterization of corneal endothelium and DM in Col8a2Q455K/Q455K (MUT) mice and human subjects. (A–F) Clinical confocal microscopy of 5-month WT endothelium (A) showing characteristic polygonal monolayer of cells with regular size and shape and 5-month MUT endothelium (B) showing increased variability in cell size and shape with DM excrescences (guttae,*) which are highly characteristic of FECD. Ten-month WT endothelium (C) showing typical cellular appearance and 10-month MUT endothelium (D) showing increased irregularity of cell size and shape with increased accumulation of guttae (*). Endothelium from a normal 54-year-old Caucasian female (E) and from a human FECD patient heterozygous for the COL8A2 Q455K mutation [F, reprinted with permission (5)] showing endothelial cell irregularities and guttae (*). Scale bar = 60 μm. (G) Central CEC density and (H) % hexagonal cells for 5-month (n= 8) and 10-month (n= 11) WT and 5-month (n= 10) and 10-month (n= 8) MUT mice. Box and whisker plots represent mean, 25th and 75th percentile values, and minimum and maximum values. (G) *P< 0.0001 compared with WT group of same age. ^P= 0.0191 compared with 5-month MUT group. (H) *P = 0.0009 for 5-month MUT and P < 0.0001 for 10-month MUT compared with WT group of same age.
Histology and electron microscopy of Col8a2 Q455K/Q455K corneal endothelium and DM
Histology of 10-month MUT and WT corneas shows comparable epithelium and stroma (Fig. 3A and C). DM guttae highly characteristic of FECD are present in MUT (Fig. 3A) and absent in WT (Fig. 3C). Higher magnification shows MUT animals with a similar DM thickness with WT (Fig. 3B and D). Transmission electron microscopy (TEM) of DM from a 5-month MUT (Fig. 3E) shows a presumably early gutta with sharp edges and internal features compatible with intracellular remnants compared with a gutta from a 10-month MUT with a more rounded, amorphous appearance (Fig. 3F). TEM of a CEC from a 5-month MUT (Fig. 3G) shows prominent, dilated rough endoplasmic reticulum (RER) compared with age-matched WT. TEM of 10-month MUT endothelium shows extremely dilated RER and abnormal mitochondrion (Fig. 3H) compared with age-matched WT.

Periodic acid-Schiff stain of formalin-fixed, paraffin-embedded sections of 10-month MUT (A and B) and WT (C and D) corneas (scale bars (A and C) = 30 μm, (B and D) = 10 μm]. (A and C) Stratified corneal epithelium (epi) at the bottom of panels and lamellar corneal stroma (str) appear comparable between MUT and WT genotypes. MUT DM shows two guttae adjacent to an endothelial cell nucleus (A, triangles). DM in MUT versus WT mice appears similar in thickness (B and D, arrows). (E–H) TEM of DM and endothelial cell from 5-month MUT (E and G) and 10-month MUT (F and H) mice [scale bars (E and F) = 2 μm, (G and H) = 500 nm]. DM gutta from a 5-month MUT (E) shows sharper, discontinuous edges with internal ultrastructural features compared with the more rounded, amorphous gutta from a 10-month MUT (F). In both (E) and (F), guttae appear to be similar in size to adjacent endothelial cells (insets). The predominant cellular ultrastructural abnormality in 5-month MUT endothelium (G) is elongated and dilated RER. In 10-month MUT endothelium (H), the RER is more severely dilated, and abnormal mitochondria (*) are present.
UPR activation in Col8a2 Q455K/Q455K corneal endothelium
Based on our report of UPR activation in end-stage FECD patient endothelium (13) and our findings of dilated RER indicative of UPR (12) in MUT mice (Fig. 3G and H), we assessed for evidence of UPR in the endothelium of MUT mice. Immunofluorescence showed increased UPR markers Grp78 and Gadd153 in 10-month MUT endothelium compared with WT (Fig. 4A). Quantitative real-time polymerase chain reaction (RT-PCR) for UPR markers showed a 2.04-fold increase in Grp78, a 2.87-fold increase in Gadd153 and a 2.26-fold increase in Atf4 messenger ribonucleic acid (mRNA) from endothelium of 12-month MUT versus WT mice (Fig. 4B). Western blotting and densitometry showed a 3.44-fold increase (0.38 ± 0.19 versus 0.11 ± 0.06, mean ± standard deviation) for Grp78 and a 2.80-fold increase (1.93 ± 0.53 versus 0.69 ± 0.71) for Gadd153 in the endothelium of 10-month MUT versus WT, whereas Col8a2 levels were similar (Fig. 4C). The comparable levels of Col8a2 protein in MUT versus WT demonstrate that the cellular effects of the Col8a2 Q455K mutation are not mediated via altered protein levels and are consistent with alternate mechanisms involving aberrant protein structure and/or function.

Assessment of UPR activation in corneal endothelium of Col8a2Q455K/Q455K (MUT) mice. (A) Immunofluorescence of UPR markers Grp78 and Gadd153 shows increased signal in the corneal endothelium (arrows) of 10-month MUT versus WT mice. (B) Quantitative RT-PCR of UPR markers Grp78, Gadd153 and Atf4 from corneal endothelium of 12-month-old MUT and WT mice. Expression was normalized to mean levels for β-actin and Gapdh, and relative expression of MUT compared with values for WT mice set to 1.0 is shown as mean ± standard deviation. *P= 0.007 for Grp78, P= 0.001 for Gadd153 and P= 0.012 for Atf4. (C) Western blot analysis with densitometry normalized to β-actin shows increased levels of Grp78 and Gadd153 and comparable levels of Col8a2 in the endothelium from 10-month MUT versus WT mice. *P= 0.03.
UPR-associated apoptosis in Col8a2 Q455K/Q455K corneal endothelium
Having established UPR activation in MUT endothelium, we assessed whether CEC death was occurring through UPR-associated apoptosis via caspase 12 and 9 activation (15,16,20). Immunofluorescence of the endothelium showed increased signal for caspase 12 in 10-month MUT versus WT and for cleaved caspase 9 in 20-month MUT versus WT mice (Fig. 5A). Quantitative RT-PCR showed a 2.97-fold increase in caspase 12 mRNA in 12-month MUT endothelium versus WT (Fig. 5B). Relative immunofluorescence intensity for cleaved caspase 9 showed a 1.81-fold increase in 20-month MUT endothelium versus WT (Fig. 5C). Terminal deoxynucleotidyl transferase -deoxyuridine, -triphosphate nick end-labeling (TUNEL) staining of 20-month MUT endothelium showed single and clustered apoptotic cells compared with no apoptosis in WT (Fig. 5D). Although these results indicate that endothelial cell loss in MUT mice is mediated by UPR-associated apoptosis via caspases 12 and 9, they do not exclude the activation of cell death mechanisms involving pathways other than the UPR.

Assessment of UPR-associated apoptosis in corneal endothelium of Col8a2Q455K/Q455K (MUT) mice. (A) Immunofluorescence of UPR-associated apoptosis markers shows increased signal in the endothelium (arrows) of 10-month (caspase 12) and 20-month (cleaved caspase 9) MUT versus WT mice. (B) Quantitative RT-PCR shows increased caspase 12 in endothelium of 12-month MUT versus WT mice (*P= 0.0003). (C) Cleaved caspase 9 immunofluorescence shows increased mean signal intensity in endothelium of 20-month MUT versus WT mice (*P= 0.0063). (D) TUNEL staining of 20-month MUT endothelium shows apoptotic cells (arrows) individually and in clusters compared with no apoptosis in WT endothelium. *TUNEL-negative nucleus of an adjacent corneal stromal keratocyte.
DISCUSSION
Our results demonstrate that knock-in of the Col8a2 Q455K mutation results in progressive CEC loss and formation of basement membrane guttae which are highly characteristic of the disease in FECD patients heterozygous for the same mutation. Our findings also establish the p.Q455K substitution as both necessary and sufficient to cause early onset FECD and provide further validation for the mouse as a powerful tool for studying human corneal endothelial diseases.
FECD is a genetically heterogeneous disease associated with mutations in multiple genes (1,5,9,10). Although the original report of FECD mutations in COL8A2 indicated 8% of probands harbored pathogenic mutations in this gene (5), a more accurate estimate of FECD patients with COL8A2 mutations is likely much lower (21). However, collagen VIII (COL8) is potentially highly relevant to the study of FECD pathogenesis as immunoelectron microscopy studies of a genetically undifferentiated series of patients demonstrated COL8 staining in abnormal areas of DM, including guttae and the posterior collagenous layer (22). Thus, it seems plausible that COL8 may play a secondary role in the pathogenesis of FECD caused by mutations in genes other than COL8A2.
Collagen VIII (COL8) is a short-chain collagen produced by CECs and is a major component of DM (23). COL8 is composed of two procollagen chains, α1 (COL8A1) and α2 (COL8A2). In vitro and in vivo studies indicate that homotrimers of both chains can be formed (24,25). Evidence also supports a heterotrimeric form of COL8 consisting of COL8A1 and COL8A2 in a 2:1 ratio (26,27). COL8A2 in humans is a 702 (698 in mice) amino acid protein consisting of two shorter, amino- and carboxy-terminal, non-collagenous domains, and a longer, central, triple-helical, collagenous domain (26). All known FECD mutations in COL8A2 occur in the triple-helical, collagenous domain. COL8A2 appears to have pleiotropic functions, and as a component of type VIII collagen has been proposed to play a role in cellular proliferation, migration and differentiation as well as providing structural support to DM (23,28–30).
Two mouse models of altered Col8a2 have been reported. Neither of these models is associated with a known COL8A2 defect in humans, and neither shows clear phenotypic similarity to FECD. The first model showed an enlarged anterior segment of the eye with thinned corneal stroma and DM in a Col8a1−/− Col8a2−/− double knock-out (28). The Col8a2−/− single knock-out showed normal anterior segment size and stromal thickness and a milder thinning of DM (28). Endothelial cell abnormalities including in situ decreased cell density and increased irregularity of cell morphology were reported for the Col8a1−/− Col8a2−/− double knock-out at an unspecified age, and a progressive nature of these changes was not indicated. In addition, these features were not reported for the Col8a2−/− single knock-out (28). Guttae and thickening of DM were reported as absent in the double knock-out (28).
The second previous mouse model of mutant Col8a2 results from a point mutation causing a glycine to aspartate substitution at amino acid 257 resulting from an ethylnitrosourea mutagenesis screen (31). This substitution produces a semi-dominant enlargement of the anterior segment and thinning of the cornea (31). The endothelium was reportedly unaltered histologically, although in situ hybridization showed reduced levels of Col8a2 expression. Furthermore, guttae characteristic of FECD was reportedly absent (31). Thus, both the Col8a2−/− and the Col8a2G257D mutant mouse lines show variable enlargement of the anterior segment and corneal thinning which phenotypically are quite different from FECD. In contrast, our Col8a2Q455K/Q455K mice show a phenotype highly similar to FECD with progressive endothelial cell morphologic changes, cell loss and characteristic DM guttae. Enlarged anterior segments and thinned corneal layers were not observed. These phenotypic differences likely reflect different pathologic mechanisms including reduced expression of Col8a2 compared with WT in the Col8a2−/− and Col8a2G257D lines, and comparable expression of mutant Col8a2 in the Col8a2Q455K/Q455K line. These differences also suggest that unlike the Col8a2−/− and Col8a2G257D lines, our model appears to be highly relevant for studying the pathogenesis of FECD resulting from mutations in COL8A2.
Despite the similar phenotypic features between the Col8a2Q455K/Q455K mice and human FECD patients with the same mutation, differences should be noted. Our mice exhibited DM guttae as was also noted in a young Q455K patient with the mildest phenotype described by Biswas et al. (5). However, ultrastructural analysis of a different patient showed a thickened DM which was not observed in our mutant mice at 10 months of age. This difference may be due to the relatively early stage disease in the mice we studied compared with the later stage disease tissue typically obtained at corneal transplantation. Thus, older mice may show more DM thickening. In addition, the Q455K mutation was also identified in a subset of patients with posterior polymorphous corneal dystrophy characterized by multi-layered ‘epithelial-like’ endothelium with desmosomal intercellular attachments and cell surface microvilli (5). No such changes were observed in our mice, thereby indicating a difference compared with humans in the effects of the Q455K mutation on endothelial cells either during development or later in life.
Biosynthesis of collagens requires the production of individual pro-α chains, post-translational prolyl and lysyl hydroxylation, and assembly into triple helices within the ER. Thus, mutations in collagens affecting protein folding or post-translational processing would be expected to induce the UPR. Several diseases caused by mutations in collagen genes result in protein misfolding and activation of the UPR, including osteogenesis imperfecta (collagen I) (32), spondyloepiphyseal dysplasia (collagen II) (33,34) and Schmid metaphyseal chondrodysplasia (collagen X) (35).
All known FECD mutations in COL8A2 produce amino acid substitutions in the central triple-helical domain of the protein and include a leucine to tryptophan change at amino acid 450 and a glutamine to lysine or valine change at amino acid 455 (5–8). These substitutions suggest different biochemical effects on the protein based on size, charge and polarity. However, it is possible that a common effect of these mutations is to alter the secondary or tertiary structure of the COL8A2 protein, thereby resulting in protein misfolding, UPR activation and ultimately UPR-associated apoptosis of endothelial cells. For the Q455K mutation in particular which changes an evolutionarily conserved neutral glutamine for a basic lysine residue, an additional mechanism leading to protein misfolding could involve hydroxylation of the aberrant lysine with subsequent glycosylation or inter-strand cross-linking resulting in misfolded COL8A2 and UPR activation.
Although this mechanism appears feasible for COL8A2 mutant FECD, the UPR also may be relevant to disease caused by mutations in other genes as well. This possibility is supported by our previous work showing UPR activation in end-stage FECD corneal specimens from genetically undifferentiated patients in whom causative COL8A2 mutations would be rare (13). In addition, a cell culture model of FECD missense mutations in the SLC4A11 gene showed aberrant protein glycosylation and accumulation in the ER, although specific markers for UPR activation were not assessed (10).
The development of a mouse model of FECD has great potential for the study of disease pathogenesis and possible treatments. Due to the decades-long disease course and absence of vision loss in milder stages, studies of early pathologic tissue have been infeasible. Instead, studies of FECD pathophysiology have used end-stage corneas obtained at transplantation. The current study utilizes corneal endothelium with a cell density of ~1240 cells/mm2 in 10-month-old mutant mice. By comparison, although perhaps with some limitations, corneas with endothelial cell loss severe enough to require transplantation have been estimated to have between 500 and 600 cells/mm2 (36,37). Thus, our present work identified UPR activation in milder FECD than has been previously studied and suggests these changes may be important in early disease pathogenesis.
Ongoing work will determine whether in vivo modification of the UPR can ameliorate the Col8a2 FECD phenotype in our model and thus provide the basis for non-surgical therapies. As suggested by previous reports (10,13), the UPR may contribute to FECD pathogenesis caused by mutations in genes other than COL8A2. If so, then modulation of the UPR may have broader impact as a potential treatment approach for FECD.
MATERIALS AND METHODS
Transgenic knock-in and animals
To generate the targeting vector, a 12.1 kb SpeI–SpeI fragment that included a portion of intron 2, the entire exon 3 and a portion of exon 4 from Col8a2 (Fig. 1A) was subcloned from mouse chromosome 4 bacterial artificial chromosome (BAC) clone 71G11 (Roswell Park Cancer Institute, Buffalo, NY, USA) into pBK-CMV vector (Agilent Technologies, Santa Clara, CA, USA) using a recombineering-based approach (19). Briefly, 500bp fragments corresponding to the 5′ and 3′ ends of the 12.1 kb SpeI–SpeI Col8a2 fragment were cloned into the pBK-CMV vector to generate the gap-repair plasmid. The gap-repair plasmid was linearized and transformed into the DY380 strain carrying the Col8a2-BAC DNA. Transformants carrying the 12.1 kb Col8a2 fragment were screened by PCR followed by DNA sequencing. The 12.1 kb Col8a2 fragment was subcloned into pBluescript (pBS, Agilent Technololgies). The C to A nucleotide change (underlined) corresponding to the p.Q455K substitution was introduced by overlap extension PCR mutagenesis using overlap primers Q455K-F (5′-GGGGCTTCCTGGG AAGCCTGGCTTGAG-3′) and Q455K-R (5′-CTCAAGCCAGGCTTCCCAGGAAGCCCC-3′). Flanking primers included KasMfeAsc-F (5′-AGCAGCGGCGCCGGTCTGCAATTGGGGTTAGGCGCGCCGCC TGGTGGTGGG ATG-3′) containing KasI, MfeI and AscI restriction enzyme sequences (underlined) and aligned with a naturally occurring KasI site within intron 3 of the mouse Col8a2 gene and 9-R (5′-CTGGTCCAGGTAGCCC TTCT-3′) located within exon 4 of Col8a2. The overlap extension product KasMfeAsc-F-9-R containing the Q455K mutation was cloned into the 12.1 kb SpeI–SpeI Col8a2 fragment at naturally occurring KasI and BsiWI sites within intron 3 and exon 4 of Col8a2. This step produced MfeI and AscI restriction sites in intron 3 which were used to insert the phospho-glycerate kinase (pGK)-neomycin-resistance selection cassette flanked by loxP sites which were PCR amplified from PL452 (kindly provided by Dr Jeremy Nathans) using primers PGKNEOMfeI-F (5′-CTCGAGCAATTGCTGCAGCCA AGCTATCGAAT-3′) and PGKNEOAscIGG-R (5′-CCGGCGCGCCACTAGTGGATCCC CTCGAGGGACCTAATAACT-3′) containing MfeI and AscI restriction sites (underlined), respectively.
The final targeting vector (Fig. 1A) was verified by restriction mapping and direct sequencing of the entire Col8a2 amino acid coding region (contained wholly within exons 3 and 4) as well as intron 3 containing the pGK-neomycin selection cassette and loxP flanking sequences. After linearization of the targeting vector using NotI, embryonic stem (ES) cell electroporation and blastocyst injection was performed by the Johns Hopkins Transgenic Mouse Core Facility.
Verification of the correct recombination event within neomycin resistant ES cell clones and subsequent progeny was performed with Southern analysis of MfeI-digested genomic DNA and a probe located within exon 4 (Fig. 1B). WT chromosomes resulted in an 18.1 kb MfeI fragment (Fig. 1A and B). Properly targeted chromosomes produced a smaller 6.1 kb MfeI fragment resulting from the insertion during targeting vector construction of an additional MfeI site within intron 3 upstream of the pGK-neomycin selection cassette (Fig. 1A and B).
Chimeric animals were bred to heterozygosity (Fig. 1B) and were then crossed with the cre-recombinase expressing line Tg(Sox2-cre)1Amc/J (Stock no. 4783, Jackson Labs, Bar Harbor, ME, kindly provided by Dr J. Nathans) resulting in excision of the pGK-neoymycin cassette flanked by loxP sites inserted into intron 3. The final correctly targeted mutant allele produced a 4.2 kb MfeI fragment (Fig. 1A and B). Sequencing of the Col8a2 gene in targeted heterozygotes demonstrated equal proportions of both the WT cytosine and MUT adenine bases specifying the p.Q455K substitution (Fig. 1C) and proper excision of the pGK-neomycin cassette. Breeding to homozygous WT or MUT genotypes was confirmed by PCR amplification, restriction enzyme digestion and agarose gel electrophoresis (Fig. 1D, see Genotyping section below). Homozygous WT and homozygous Col8a2Q455K/Q455K (MUT) mice at the specified ages were used in subsequent studies.
All procedures were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Johns Hopkins Medical Institutions.
Genotyping
DNA was extracted from mouse tails (Qiagen DNeasy Blood and Tissue Kit, Valencia, CA, USA). Genotyping for Col8a2Q455K/Q455K mutants was performed by PCR amplification of a 574 bp fragment of the Col8a2 gene using the forward primer 5′-ATTCGA GGAGACCAAGGGCCTAAT-3′ and reverse primer 5′-AAGTGAGCACTGCAGTAA AGG CTG-3′. PCR products were cleaned (Fermentas GeneJET PCR Purification Kit, Glen Burnie, MD, USA), digested with Fnu4HI restriction enzyme (New England BioLabs, Ipswitch, MA, USA) and subjected to agarose gel electrophoresis. Mutant containing PCR fragments are undigested by Fnu4HI, whereas WT fragments are digested into 384 bp and 190 bp fragments (Fig. 1D).
Clinical confocal microscopy
Immediately after euthanasia, 1% sodium hyaluronate viscoelastic (Abbott Medical Optics, Abbott Park, IL, USA) was injected into the orbit to prolapse the globe, and whiskers were trimmed. Mice were positioned on a specialized platform adapted to a commercially available clinical confocal microscope (Nidek Confoscan 3, Fremont, CA, USA). Endothelial cell imaging and quantitative analysis were performed using the Confoscan 3 software. For the normal human subject, informed consent was obtained and the corneal endothelium was imaged using a Nidek Confoscan 4 instrument.
Histology
Whole eyes were placed in Excalibur's Alcoholic Z-Fixative and sent to Excalibur Pathology (Moore, OK, USA). Eyes were embedded in paraffin, and 5 μm sections were stained with periodic acid-Schiff/Gill III hematoxylin (StatLab Medical, McKinney, TX, USA).
Transmission electron microscopy
Whole globes were enucleated and fixed in 2.5% glutaldehyde/2% paraformaldehyde, pH 7.4, overnight at 4°C. Corneas were excised and bisected. Corneal halves were then washed in 0.1 m phosphate buffer followed by a 2 h, room-temperature fixation in 1% osmium tetroxide in 0.1 m phosphate buffer, pH 7.4. The specimens were then washed and dehydrated in a series of 50–100% ethanol followed by a 1 h fixation in 1% uranyl acetate in 100% ethanol at room temperature in the dark. Post-dehydration, specimens were washed twice in propylene oxide for 15 min each and incubated in a 1:1 mixture of propylene oxide to LX-112 resin (Ladd Research, Williston, VT, USA) overnight at room temperature. Specimens were submerged in 100% resin, subjected to vacuum for 5 h before embedding in freshly made LX-112 resin, and polymerized at 60°C for 2 days before sectioning. Sixty-eight nanometer ultrathin sections were cut with a Leica Ultramicrotome UCT (Wetzlar, Germany) and imaged using a Hitachi H7600 transmission electron microscope (Pleasanton, CA, USA).
Immunofluorescence microscopy
All immunofluorescence studies included one cornea from three different Col8a2Q455K/Q455K mutant (n= 3) animals and three different WT (n= 3) animals. Whole corneas were dissected and fixed for 1 h at room temperature in 4% paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4. Specimens were then incubated overnight in 20% sucrose at 4°C before embedding in Tissue-Tek O.C.T. (Sakura, Torrance, CA, USA) with dry ice chilled methyl-butane. Eight micrometer sections were taken from the central cornea and mounted on Superfrost Plus Slides (Fisher Scientific, Pittsburgh, PA, USA). Sections were dried overnight and permeabilized with pre-cooled absolute methanol at −20°C for 5 min. Slides were blocked in PBS containing 5% goat serum (Sigma, St Louis, MO, USA) and 0.3% Triton X-100 (Sigma) for 30 min. This was followed by incubation with primary antibodies for 90–120 min and secondary antibody for 30–45 min. All antibodies were diluted in PBS containing 1% bovine serum albumin (Sigma) and 0.3% Triton X-100. Antibodies used were: Grp78 (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Gadd153 (1:100, Santa Cruz Biotechnology), cleaved caspase 9 (1:50, Cell Signaling Technology, Danvers, MA), caspase 12 (1:300, Abcam, Cambridge, MA, USA) and Alexa Fluor 488 goat anti-rabbit IgG [Cell Signalling, concentration when used as secondary antibody with Grp78 (1:500), Gadd153 (1:200), cleaved caspase 9 (1:500), caspase 12 (1:1000)]. Sections were counter-stained with 4’,6-diamidino-2-phenylindole (DAPI) (Roche Applied Science, Indianapolis, IN, USA) and mounted in Fluorescent Mounting Medium (DAKO, Carpinteria, CA, USA).
Cleaved caspase 9 fluorescence quantification was performed on three randomly selected, non-overlapping fields for a central corneal section from each animal. Image analysis was performed using NIH ImageJ (http://imagej.nih.gov/ij/download/). Fluorescence intensity for each animal was obtained by taking the difference of the endothelial fluorescence for each image from the background fluorescence taken from the same-sized area of the adjacent corneal stroma. Values obtained from the three separate non-overlapping fields were then averaged together to obtain the average fluorescence intensity for a single Col8a2Q455K/Q455K mutant or WT animal. One cornea was used from three different mutant (n= 3) and three different WT (n= 3) mice.
Terminal deoxynucleotidyl TUNEL staining for apoptosis was performed using the in situ cell death detection kit, TMR red (Roche Applied Science). Sections were counterstained with DAPI (Roche Applied Science) and mounted in Fluorescent Mounting Medium (DAKO).
All imaging was performed on an LSM 510 META confocal microscope using a ×20 objective and ×3 digital zoom (Zeiss, Thornwell, NY, USA).
Western blotting and quantification
DM and endothelial cells were stripped from freshly dissected, 10-month-old Col8a2Q455K/Q455K mutant and WT mouse corneas and homogenized in Tissue Protein Extract Reagent (Thermo Fisher Scientific, Rockford, IL, USA) with 1% protease inhibitor cocktail (Sigma) and 1% ethylenediaminetetraacetic acid (Sigma). Each sample contained both corneas of the same MUT (n= 4) or WT (n= 4) animal. Four MUT and four WT animals were used. The mixture was then micro-centrifuged at 4°C for 10 min at 12 000 rpm. The lysate was removed and the protein concentration was quantified by BCA Protein Assay Kit (Thermo Fisher Scientific). Eight micrograms of protein was mixed with 10 μl of 4× loading dye (NuPage, Invitrogen, Carlsbad, CA, USA) with 2-mercaptoethanol (Sigma) and heated at 65°C for 5 min. Samples were loaded onto a 10% Tris–HCl Ready Gel (BioRad, Hercules, CA, USA) and subjected to sodium dodecylsulfate–polyacrylamide gel electrophoresis separation for 1 h at 120 V. Proteins were transferred to a polyvinylidene fluoride membrane (BioRad) and incubated in blocking buffer made of 5% non-fat milk in PBS with 0.1% Tween-20. Membranes were then incubated in primary antibodies: Grp78 (1:500, Cell Signaling Technology), Gadd153 (1:1000, Santa Cruz Biotechnology) and Col8a2 (1:2000, kindly provided by Dr Paul Davis) diluted in blocking buffer for 1 h at room temperature. Subsequently, membranes were washed and incubated in 1:10 000 dilution of anti-rabbit IgG, horseradish peroxidase conjugated antibody (GE Healthcare, Piscataway, NJ, USA) diluted in blocking buffer for 45 min at room temperature. Loading controls were assayed by probing with β-actin (1:2000, Cell Signaling Technology) as primary antibody after stripping with Restore Stripping Buffer (Thermo Fisher Scientific). Low abundance proteins were detected using SuperSignal West Dura (Thermo Fisher Scientific) and higher abundance proteins were detected using HyGlo Quick Spray (Denville Scientific, Metuchen, NJ, USA). Densitometry analysis was performed using Adobe Photoshop CS5 as previously described (http://www.lukemiller.org/journal/2007/08/quantifying-western-blots-without.html).
Quantitative RT–PCR
Total RNA was extracted from murine corneal endothelium on stripped DMs (four eyes per group) using TRIzol reagent (Invitrogen) followed by RNeasy column (Qiagen) purification. Complementary DNA was generated using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) and pre-amplified using the TaqMan PreAmp Master Mix (Applied Biosystems). Assays were performed in triplicate on a total of three groups [2 animals (4 corneas)/group] for each genotype using a StepOne Plus Cycler (Applied Biosystems) and Universal Master Mix II, no UNG (Applied Biosystems). Taqman inventoried gene expression assays (Applied Biosystems) used were Mm01333323_g1 (Grp78), Mm00492097_m1 (Gadd153), Mm00515324_m1 (Atf4), Mm00438038_m1 (Caspase 12), Mm00607939_s1 (β-actin) and Mm99999915_g1 (Gapdh). The relative gene expression in Col8a2Q455K/Q455K versus WT endothelium was normalized to housekeeping genes β-actin and Gapdh according to the comparative CT method (38).
Statistical analysis
Unpaired, two-tailed, t-tests were performed using PRISM 4 software (Graphpad Software, La Jolla, CA, USA). P-values < 0.05 were considered statistically significant.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the National Institutes of Health (grant numbers EY15523; to A.S.J., EY019874; to A.S.J., EY001765 to Wilmer Microscopy Core Facility); Research to Prevent Blindness (Career Development Award to A.S.J., Special Scholar Award to A.S.J.); Wilmer Professors Fund to (A.S.J.); Medical Illness Counseling Center to (A.S.J.) and Deutsche Forschungsgemeinschaft (German Research Foundation) (grant number DFG/MA 5110/2-1 to M.M.).
ACKNOWLEDGEMENTS
The authors thank Jeremy Nathans, MD, PhD, David Ginty, PhD and Paul Davis, PhD, for technical assistance and the NIHR Manchester Biomedical Research Centre.
REFERENCES
Articles from Human Molecular Genetics are provided here courtesy of Oxford University Press
Full text links
Read article at publisher's site: https://doi.org/10.1093/hmg/ddr473
Read article for free, from open access legal sources, via Unpaywall:
https://academic.oup.com/hmg/article-pdf/21/2/384/1911782/ddr473.pdf

Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Smart citations by scite.ai
Explore citation contexts and check if this article has been
supported or disputed.
https://scite.ai/reports/10.1093/hmg/ddr473
Article citations
[Pathogenesis of Fuchs endothelial corneal dystrophy, the fibrillar layer and individualized treatment].
Ophthalmologie, 121(10):787-795, 01 Oct 2024
Cited by: 0 articles | PMID: 39352514
Characterization of a Novel Mouse Model for Fuchs Endothelial Corneal Dystrophy.
Invest Ophthalmol Vis Sci, 65(4):18, 01 Apr 2024
Cited by: 0 articles | PMID: 38587441 | PMCID: PMC11005065
Endoplasmic Reticulum Stress Disrupts Mitochondrial Bioenergetics, Dynamics and Causes Corneal Endothelial Cell Apoptosis.
Invest Ophthalmol Vis Sci, 64(14):18, 01 Nov 2023
Cited by: 2 articles | PMID: 37962528 | PMCID: PMC10653263
Squishy matters - Corneal mechanobiology in health and disease.
Prog Retin Eye Res, 99:101234, 02 Jan 2024
Cited by: 1 article | PMID: 38176611 | PMCID: PMC11193890
Review
 Free full text in Europe PMC
Free full text in Europe PMCAnimal Models in Eye Research: Focus on Corneal Pathologies.
Int J Mol Sci, 24(23):16661, 23 Nov 2023
Cited by: 6 articles | PMID: 38068983 | PMCID: PMC10706114
Review
Free full text in Europe PMC
Go to all (69) article citations
Data
Data behind the article
This data has been text mined from the article, or deposited into data resources.
BioStudies: supplemental material and supporting data
Diseases
- (1 citation) OMIM - 136800
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy.
Invest Ophthalmol Vis Sci, 54(3):1887-1897, 28 Mar 2013
Cited by: 45 articles | PMID: 23422828 | PMCID: PMC3626522
Biomechanical changes to Descemet's membrane precede endothelial cell loss in an early-onset murine model of Fuchs endothelial corneal dystrophy.
Exp Eye Res, 180:18-22, 22 Nov 2018
Cited by: 13 articles | PMID: 30471280 | PMCID: PMC6389414
Endothelial Cdkn1a (p21) overexpression and accelerated senescence in a mouse model of Fuchs endothelial corneal dystrophy.
Invest Ophthalmol Vis Sci, 53(10):6718-6727, 28 Sep 2012
Cited by: 22 articles | PMID: 22956607 | PMCID: PMC3462481
Mitochondrial Dysfunction and Mitophagy in Fuchs Endothelial Corneal Dystrophy.
Cells, 10(8):1888, 26 Jul 2021
Cited by: 21 articles | PMID: 34440658 | PMCID: PMC8392447
Review
Free full text in Europe PMC
Funding
Funders who supported this work.
NEI NIH HHS (3)
Grant ID: EY001765
Grant ID: EY019874
Grant ID: EY15523


