Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data.
![]() Free full text in Europe PMC
Free full text in Europe PMC
Abstract
Background
Functional genomics studies are yielding information about regulatory processes in the cell at an unprecedented scale. In the yeast S. cerevisiae, DNA microarrays have not only been used to measure the mRNA abundance for all genes under a variety of conditions but also to determine the occupancy of all promoter regions by a large number of transcription factors. The challenge is to extract useful information about the global regulatory network from these data.Results
We present MA-Networker, an algorithm that combines microarray data for mRNA expression and transcription factor occupancy to define the regulatory network of the cell. Multivariate regression analysis is used to infer the activity of each transcription factor, and the correlation across different conditions between this activity and the mRNA expression of a gene is interpreted as regulatory coupling strength. Applying our method to S. cerevisiae, we find that, on average, 58% of the genes whose promoter region is bound by a transcription factor are true regulatory targets. These results are validated by an analysis of enrichment for functional annotation, response for transcription factor deletion, and over-representation of cis-regulatory motifs. We are able to assign directionality to transcription factors that control divergently transcribed genes sharing the same promoter region. Finally, we identify an intrinsic limitation of transcription factor deletion experiments related to the combinatorial nature of transcriptional control, to which our approach provides an alternative.Conclusion
Our reliable classification of ChIP positives into functional and non-functional TF targets based on their expression pattern across a wide range of conditions provides a starting point for identifying the unknown sequence features in non-coding DNA that directly or indirectly determine the context dependence of transcription factor action. Complete analysis results are available for browsing or download at http://bussemaker.bio.columbia.edu/papers/MA-Networker/.Free full text
Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data
 1,2
1,2Abstract
Background
Functional genomics studies are yielding information about regulatory processes in the cell at an unprecedented scale. In the yeast S. cerevisiae, DNA microarrays have not only been used to measure the mRNA abundance for all genes under a variety of conditions but also to determine the occupancy of all promoter regions by a large number of transcription factors. The challenge is to extract useful information about the global regulatory network from these data.
Results
We present MA-Networker, an algorithm that combines microarray data for mRNA expression and transcription factor occupancy to define the regulatory network of the cell. Multivariate regression analysis is used to infer the activity of each transcription factor, and the correlation across different conditions between this activity and the mRNA expression of a gene is interpreted as regulatory coupling strength. Applying our method to S. cerevisiae, we find that, on average, 58% of the genes whose promoter region is bound by a transcription factor are true regulatory targets. These results are validated by an analysis of enrichment for functional annotation, response for transcription factor deletion, and over-representation of cis-regulatory motifs. We are able to assign directionality to transcription factors that control divergently transcribed genes sharing the same promoter region. Finally, we identify an intrinsic limitation of transcription factor deletion experiments related to the combinatorial nature of transcriptional control, to which our approach provides an alternative.
Conclusion
Our reliable classification of ChIP positives into functional and non-functional TF targets based on their expression pattern across a wide range of conditions provides a starting point for identifying the unknown sequence features in non-coding DNA that directly or indirectly determine the context dependence of transcription factor action. Complete analysis results are available for browsing or download at http://bussemaker.bio.columbia.edu/papers/MA-Networker/.
Background
For various organisms, DNA microarrays have been used to measure the mRNA abundance for essentially all protein-coding genes in the genome under a large number of conditions [1,2]. Microarray technology can also be combined with chromatin-immunoprecipitation (ChIP) or chromatin profiling (DamID) to quantify the occupancy of upstream non-coding regions by transcription factors or other chromatin-associated proteins [3-9]. In the budding yeast Saccharomyces cerevisiae, ChIP has been used to globally map the binding sites of over a hundred transcription factors [4]. Moreover, mRNA expression data for over a thousand conditions has been published. The challenge is to find new ways to extract knowledge about the regulatory mechanisms that govern the cell by combining these different types of data [10-14].
Initiation of transcription in eukaryotes is a complicated process that depends on the binding of transcription factors (TFs) and chromatin-modifying enzymes to the promoter region as well as the recruitment of the RNA Polymerase II complex to the transcription start site. Transcriptional control is combinatorial, and cooperative binding of multiple factors on the same promoter region and/or cooperative recruitment of the Pol II complex is often required for transcriptional activation [15]. Occupancy of the promoter region of a gene by a transcription factor is thus a necessary but not a sufficient condition for the gene to be controlled by it. As a consequence, genome-wide transcription factor binding patterns measured using ChIP or DamID microarray experiments alone can only indicate the potential for a gene to be regulated by a given TF. Independent information will be required to establish that the gene is indeed a functional target of the factor.
Deletion or over-expression of a transcription factor, combined with genomewide microarray profiling of the difference in expression between mutant and wild type, is also widely used to infer regulatory interactions. However, drastic perturbation of the genetic network outside the physiologically relevant range may lead to false target prediction, or the mutant strain may simply not be viable. Moreover, direct and indirect targets of the factor cannot be distinguished using this approach.
When mRNA abundances for all genes are compared between two experimental conditions in a microarray experiment, the observed differential expression pattern is usually a superposition of responses of various pathways, mediated by signaling cascades that end at the level of transcription factors. It has previously been shown that these changes in TF activity can be quantitatively inferred by performing multivariate regression analysis on the expression log-ratios from a single microarray experiment [16-19]. Transcription factors are implicitly represented by a consensus motif for their DNA binding sites, and the regression coefficients estimate the changes in TF activity.
In the present study we will instead use ChIP occupancy log-ratios as predictors for expression. No sequence information will be used, and it is therefore not necessary that a DNA consensus motif be known for the TFs. Again, multivariate regression analysis of a single genomewide set of mRNA log-ratios on the genomewide binding profiles of a large number of TFs for which ChIP data is available can be used to quantify to what extent each transcription factor is responsible for the observed changes in mRNA expression.
When the regression procedure is performed in parallel on a large library of expression data, the inferred changes in TF activity for each comparison can be combined into a transcription factor activity profile (TFAP) for each transcription factor ("Step 1" in Fig. Fig.1a).1a). Each TFAP represents a highly specific regulatory signature, as is shown for three transcription factors in Fig. Fig.1b:1b: The activity of the G2 phase related factor Ndd1p oscillates during the cell cycle (blue), but shows little or no response to nutrient depletion and other stress conditions (red), or changes in alpha pheromone concentration (green). Complementary behavior is seen for the TCA cycle regulator Hap4p and the mating-related factor Ste12p.

(a) Overview of our method for determining regulatory coupling strengths between transcription factors and their putative target genes. Inputs are (i) a library of microarray expression data for a large number of conditions and (ii) genomewide (ChIP) occupancy data for one or more transcription factors. In the first step of our algorithm, a matrix of transcription factor activities is inferred by using regression analysis to explain the mRNA expression pattern under each condition in terms of the ChIP data for each transcription factor. In the second step, a matrix of regulatory coupling strengths is determined by computing the correlation between each transcription factor activity profile (TFAP) and the mRNA expression profile of each gene. (b) Examples of transcription factor activity profiles. The activity profiles of three transcription factors (Hap4, Ndd1, Ste12) are shown across stress response, pheromone response, and cell cycle [28-30]. Significant changes in activity of the TCA cycle regulator Hap4p occur mostly in metabolic stress conditions, while changes in the activity of the cell cycle regulator Ndd1p and the pheromone-dependent regulator Ste12p are associated with the cell cycle and signal transduction experiments, respectively. (c) Examples of scatter plots of ChIP binding log-ratio versus coupling factor. In the scatter plots, black dots denote unbound (B-) genes, red dots denote bound and coupled genes (B+/C+), while green dots denote genes that are bound but not coupled (B+/C-). A threshold of P = 10-3 was used to determine significant binding as described in Lee et al. [4]. A threshold for coupling was determined by requiring a false discovery rate of 5%, as described in Methods.
One expects the mRNA expression profile of a gene regulated by a specific transcription factor to be similar to the TFAP of that factor. We therefore investigated whether the linear correlation across the experiment library between a TFAP and the mRNA expression profile of a gene whose promoter is bound by the factor could be interpreted as a regulatory coupling strength and used to improve the specificity of target prediction. To this end, we constructed a matrix of regulatory coupling strengths between all transcription factors and all genes ("Step 2" in Fig. Fig.1a).1a). When this information is combined with the original ChIP data for a given TF, the ChIP log-ratio and coupling strength for each gene can be shown simultaneously in a 2D scatter plot (Fig. (Fig.1c).1c). The fact that each gene has two parameters associated with it allows a more sophisticated classification than is possible based on ChIP alone. We first defined a set "B+" of genes that are significantly bound by a TF (we required the P-value reported by Lee et al. to be smaller than 10-3) [4]. Next, we then partitioned the "B+" gene set into two subsets "B+/C+" and "B+/C-" based on whether or not their mRNA expression profile was significantly correlated with the TFAP (Pearson correlation, 5% false discovery rate). Our hypothesis is that the B+/C+ genes (shown in red in Fig. Fig.1c)1c) are the functional direct targets of the factor, while the binding to the promoter region of the B+/C- genes (shown in green) is non-functional.
Results and discussion
Only a subset of genes bound by each TF is controlled by it
We have focused on the yeast Saccharomyces cerevisiae, for which a wealth of functional genomics data is available. We compiled a library of ~750 expression patterns from various sources, and combined it with the genome-wide promoter occupancies in mid-log phase and rich medium for 113 TFs as measured by Lee et al. [4]. It was determined that 37 transcription factor occupancy patterns out of the full set of 113 are significant predictors of mRNA expression for one or more experiments in our library (see Methods). Note that the library of expression data we used obviously does not cover all possible experimental conditions and our method is therefore likely to underestimate the number of transcription factors that are present in the nucleus under the conditions used by Lee et al. [4]. For each of the 37 factors selected for further analysis, a transcription factor activity profile (TFAP) was computed ("Step 1" in Fig. Fig.1a)1a) and the TF-target coupling strength was determined ("Step 2" in Fig. Fig.1a).1a). The number of genes in each group (B-, B+/C+, and B+/C-) is listed in Table Table11 for the 37 transcription factors analyzed. On average 58% of significantly bound genes are classified as significantly coupling genes. Activity profiles and B+/C+ target predictions for all 37 factors are available on the website supporting this paper.
Table 1
Classification of genes according to ChIP data and inferred regulatory coupling.
| TF | B- | B+/C+ | B+/C- | Unclassified* |
| Abf1 | 5638 | 138 | 136 | 470 |
| Ace2 | 5843 | 33 | 33 | 473 |
| Arg81 | 5985 | 11 | 10 | 376 |
| Bas1 | 5975 | 28 | 13 | 366 |
| Cad1 | 5854 | 26 | 13 | 489 |
| Dal81 | 5823 | 24 | 16 | 519 |
| Dig1 | 5872 | 20 | 11 | 479 |
| Fhl1 | 5754 | 146 | 37 | 445 |
| Fkh2 | 5261 | 61 | 45 | 1015 |
| Gal4 | 5149 | 20 | 20 | 1193 |
| Gat3 | 5891 | 40 | 29 | 422 |
| Gcn4 | 5919 | 58 | 21 | 384 |
| Hal9 | 5948 | 4 | 13 | 417 |
| Hap4 | 5939 | 47 | 21 | 375 |
| Hir1 | 5963 | 19 | 10 | 390 |
| Hir2 | 5932 | 8 | 13 | 429 |
| Hsf1 | 5929 | 35 | 17 | 401 |
| Leu3 | 5988 | 8 | 13 | 373 |
| Mbp1 | 5641 | 65 | 40 | 636 |
| Mcm1 | 5709 | 42 | 46 | 585 |
| Met31 | 5983 | 15 | 13 | 371 |
| Msn4 | 5952 | 23 | 7 | 400 |
| Mss11 | 4735 | 11 | 9 | 1627 |
| Ndd1 | 5799 | 50 | 42 | 491 |
| Nrg1 | 5912 | 61 | 20 | 389 |
| Rlm1 | 5900 | 13 | 24 | 445 |
| Sig1 | 5683 | 0 | 0 | 699 |
| Sko1 | 5155 | 2 | 0 | 1225 |
| Sok2 | 4881 | 10 | 6 | 1485 |
| Stb1 | 5791 | 13 | 10 | 568 |
| Ste12 | 5725 | 19 | 31 | 607 |
| Sum1 | 5919 | 35 | 26 | 402 |
| Swi4 | 5528 | 75 | 48 | 731 |
| Swi5 | 5358 | 47 | 45 | 932 |
| Thi2 | 5940 | 5 | 1 | 436 |
| Yap1 | 5959 | 28 | 16 | 379 |
| Yap6 | 5924 | 33 | 54 | 371 |
*Genes whose expression level in microarray data or binding P-value in Lee et al. is not available. The number of genes in each of the categories (B-, unbound genes; B+/C+, bound and coupling genes; B+/C-, genes that are bound but do not couple) is shown for each of the 37 transcription factors analyzed. On average, 58% of the significantly bound genes were classified in the B+/C+ group.
Enrichment for specific functional categories
Several analyses were performed to validate our results. First, we established that B+/C+ genes are significantly enriched for specific Gene Ontology (GO) categories (hypergeometric distribution; 5% false discovery rate) [20]. This result is not surprising, as we would already expect the set B+ of ChIP positives per se to be enriched for roughly the same functional categories. By contrast, for almost all TFs analyzed we found no significant enrichment for any GO category in the set of non-coupling (B+/C-) genes (Fig. (Fig.2,2, see supplementary website for details). This result is very significant because it suggests that our criterion for distinguishing functional from non-functional TF targets based on regulatory coupling is accurate: There seems to have been no evolutionary pressure on the set of B+/C- genes.

Enrichment for functional annotation. The number of significantly over-represented Gene Ontology (GO) categories in the group B+/C+ of genes that couple to transcription factor activity (red) and the group B+/C- of genes that do not couple (green) for each of the 37 transcription factors analyzed. No significant enrichment for any GO category was found in most B+/C- gene groups, supporting the hypothesis that only the coupling B+/C+ genes are functional targets.
Transcriptional response to transcription factor deletion
Next, we analyzed the expression response to transcription factor deletion for the B+/C+ and B+/C- genes. The mean and the standard deviation of the gene expression log-ratio between mutant and wild type as obtained in Hughes et al. were calculated for all genes in the genome, as well as for the B+/C+ and B+/C- groups [21]. A sample t-test was performed to determine the significance of the change in expression. The B+/C+ genes show a significant change in mRNA expression for the 7 transcription factors for which deletion and ChIP data are both available. By contrast, the response of the B+/C- genes is insignificant for most transcription factors (Table (Table22).
Table 2
Analysis of transcriptional response to transcription factor deletion.
| Transcription Factor | Genomewide | B+/C+ | B+/C- | |||||
| Mean | SD | Mean | SD | -log10(p) | Mean | SD | -log10(p) | |
| Dig1 | 0.759 | 0.198 | 0.567 | 0.360 | 3.76 | 0.846 | 0.080 | 0.04 |
| Gcn4 | 0.785 | 0.177 | 0.294 | 0.329 | 28.23 | 0.793 | 0.170 | 0.24 |
| Hir2 | 0.724 | 0.218 | 0.180 | 0.295 | 4.00 | 0.760 | 0.194 | 0.14 |
| Mbp1 | 0.830 | 0.121 | 0.541 | 0.324 | 27.86 | 0.770 | 0.172 | 2.79 |
| Swi4 | 0.524 | 0.338 | 0.348 | 0.352 | 4.89 | 0.533 | 0.333 | 0.24 |
| Swi5 | 0.744 | 0.204 | 0.583 | 0.373 | 5.98 | 0.759 | 0.186 | 0.17 |
| Yap1 | 0.756 | 0.201 | 0.471 | 0.376 | 7.66 | 0.587 | 0.337 | 2.67 |
The mean and the standard deviation of gene expression log-ratio between mutant and wild type as obtained in Hughes et al. were calculated for all genes in the genome as well as for the B+/C+ and B+/C- groups [21]. A sample t-test was performed to determine the significance of the change in expression. The B+/C+ genes show a significant change in mRNA expression for the 7 transcription factors for which deletion and ChIP data is available. By contrast, the response of the B+/C- genes is insignificant for most transcription factors.
Enrichment of promoter regions for consensus binding motifs
In a third analysis to validate our results, we tested for over-representation of 4 different cell cycle related DNA consensus motifs (MCB, SCB, Swi5p and SFF) in the upstream regions of 7 cell cycle related TFs (Ace2, Fkh2, Mbp1, Mcm1, Ndd1, Swi4 and Swi5). The binomial distribution was used to score motif enrichment in the B+/C+ and B+/C- gene sets for each of the transcription factors. We found certain DNA motifs to be significantly over-represented in B+/C+ genes for one or more TFs, but dramatically less so in B+/C- genes (Table (Table3).3). Finally, we defined B+/C+ groups using duplicate ChIP experiments for 7 cell cycle regulators performed by Simon et al. and found the overlap with the B+/C+ genes for the data of Lee et al. to be 85% on average [4,22].
Table 3
Over-representation of four cell cycle related motifs.
| Motif | Ace2 | Fkh2 | Mbp1 | Mcm1 | Ndd1 | Swi4 | Swi5 | |||||||
| B+/C+ | B+/C- | B+/C+ | B+/C- | B+/C+ | B+/C- | B+/C+ | B+/C- | B+/C+ | B+/C- | B+/C+ | B+/C- | B+/C+ | B+/C- | |
| ACGCGT (MCB) | 38.4 | 8.3 | 8.2 | 0.0 | ||||||||||
| CGCGAAA (SCB) | 7.0 | 1.3 | 19.1 | 2.8 | ||||||||||
| AACCAGC (Swi5p) | 5.1 | 0.6 | 2.6 | 0.8 | ||||||||||
| GTAAACA (SFF) | 12.9 | 1.8 | 3.1 | 1.4 | 8.3 | 0.6 | 3.5 | 0.1 | ||||||
The binomial distribution was used to score motif enrichment in the B+/C+ and B+/C- gene sets for seven cell cycle related transcription factors. The value of -log10(P) is shown only for those combinations of motifs and factors where the motif was significantly overrepresented among the genes bound by the factor. In most cases, the motif is not over-represented in the B+/C- gene group, and in all cases, the over-representation among B+/C- genes is far less significant than among B+/C+ genes.
Assigning directionality to divergently transcribed promoters
Taken together, the results mentioned above convincingly demonstrate that the use of a coupling factor threshold as a novel additional criterion leads to significantly improved specificity in the prediction of functional TF targets. The biological implications of our analysis are highlighted in the case of divergently transcribed genes that share a common promoter region, represented as a single microarray probe. There are 1592 such probes out of the total 4532 probes in the ChIP experiments of Lee et al. [4]. When the ChIP data indicate that a TF binds to the intergenic region, nothing can be said about whether it regulates one of the genes or both based on that information alone. By contrast, our regulatory coupling analysis naturally allows us to distinguish between these different scenarios and make precise statements about which genes are controlled by each of the factors that occupy the promoter region (see Fig. Fig.3).3). Both uni- and bi-directional control by TFs is observed. Indeed, we found the functional annotation of the protein encoded by the coupled targets to be consistent with what was known about the function of the bound TF in most cases analyzed [20].

Assigning directionality to divergently transcribed promoters. For pairs of divergently transcribed genes sharing a single promoter region occupied by one or more transcription factors, our method can be used to determine which gene is regulated by which factor. In the diagrams, genes are represented as squares with arrows showing the transcription direction; transcription factors are shown as ovals. The numbers shown are significance scores for the coupling between the transcription factor and the gene, equal to the negative 10-based logarithm of the P-value. Significant regulatory relationships are shown as arrows to colored boxes. In (A), the cell cycle transcription factor Mbp1p regulates the recombinase RAD51 but not the endopeptidase PUP3, while in (B), the putative rRNA processing regulator Fhl1p regulates the ribosomal subunit RPL40B but not the protein kinase MLP1. In the scenario illustrated in (C), both Nrg1p and Hap6p bind to the intergenic upstream region of FSP2 and HXT9. The coupling analysis shows that Nrg1p in this case works bi-directionally and regulates both genes, while Hap6p regulates neither gene.
Revealing intrinsic limitations of TF deletion experiments
Since TF occupancy data from ChIP experiments can be used to separate direct from indirect targets among the genes that respond to TF deletion, combining ChIP data with deletion data can potentially achieve the same goal as our more sophisticated analysis. Keeping Occam's Razor in mind, it is therefore important to investigate to what extent mRNA expression log-ratios from a TF deletion mutant vs. wild type experiment can replace our regulatory coupling strength on the vertical axis in Fig. Fig.1b.1b. We defined sets B+/D+ and B+/D- for each of the seven TFs for which data are available in Hughes et al., the D+ genes being those that show a response to the TF deletion at P < 10-2, using the P-value provided by those authors [21]. In the regulatory coupling analysis described above, we found no significant enrichment for specific GO categories in the group B+/C- of genes that are bound but not coupled. In the present case however, similar levels of enrichment for GO categories are found for B+/D+ and B+/D- genes (Table (Table4).4). This result indicates that a substantial fraction of the functional direct targets of a typical transcription factor is being missed even if one combines transcription factor deletion data with ChIP data. A plausible explanation for this lack of sensitivity is that each TF deletion experiment, by definition, is performed under a single condition in which not all possible co-factors of the deleted TF may be present. By using a large and heterogeneous library of experimental conditions as input, our method samples most or all co-factor combinations that occur as the context for control by each TF, naturally taking into account the combinatorial nature of transcriptional control.
Table 4
Replacing regulatory coupling strength by response to transcription factor deletion.
| Transcription Factor | Number of genes in each group | GO category | ||||
| B- | B+/D+ | B+/D- | Not available | B+/D+ | B+/D- | |
| Dig1 | 5881 | 1 | 30 | 115 | 32 | 16 |
| Gcn4 | 5928 | 16 | 63 | 60 | 19 | 17 |
| Hir2 | 5941 | 2 | 19 | 105 | 24 | 5 |
| Mbp1 | 5647 | 2 | 103 | 315 | 22 | 12 |
| Swi4 | 5535 | 19 | 106 | 407 | 9 | 6 |
| Swi5 | 5368 | 10 | 82 | 607 | 2 | 0 |
| Yap1 | 5968 | 7 | 37 | 55 | 0 | 0 |
We analyzed the performance of a scheme in which our TF-gene coupling factor was replaced by the change in mRNA abundance in response to transcription factor deletion as a predictor of true targets. Significant binding (B+) was defined as before, while a significant response to deletion (D+) required P < 10-2. The number of significantly over-represented GO categories is listed for both the B+/D+ and the B+/D- gene groups. The results indicate that TF deletion data is less useful than our coupling factor for distinguishing functional target genes from non-functional ones.
Conclusions
Our results underscore the unique added value of ChIP data such as that of Lee et al. when it is used in combination with a library of mRNA expression data [4]. We found that roughly half of the transcription factor targets predicted by ChIP are nonfunctional. Although some of these will be false positives of the ChIP technology, especially for TFs that are not present in active form in the nucleus under the conditions used by Lee et al., we believe that our results instead point to interesting biology: TF binding can fail to confer transcription of a nearby gene for a variety of reasons, including competition with nearby activators or repressors, local or global chromatin conformation, or lack of partners for cooperative recruitment of the Pol II complex.
Several works have relied on representing a TF by its mRNA expression profile in order to discover connections between transcription factors and their targets [23-25]. By contrast, our method infers changes in TF activity by analyzing the mRNA levels of putative TF targets. This allows us to analyze regulatory relationships even if the TF is modulated in a purely post-translational manner, e.g. by phosphorylation. The reliable classification of ChIP positives into functional and non-functional TF targets, as it has been presented here, provides a starting point for future research aimed at identifying the unknown sequence features in non-coding DNA that directly or indirectly determine the context dependence of TF action.
Methods
Microarray expression and binding data
A library of 751 genomewide mRNA expression patterns (transcriptomes) was compiled from a variety of sources (see supplementary data for complete references). ChIP data for 113 transcription factors was downloaded from the website accompanying Lee et al. [4]. We used the P-values provided by these authors to determine which genes were significantly bound by each given factor at a confidence level of P < 10-3. All microarray data used in our analysis was represented as log-ratio base two.
Transcription factor activity profiles
For each separate transcriptome t, we used the following multivariate regression model to infer transcription factor activities for each microarray experiment:
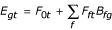
Here Egt represents the mRNA expression log-ratio of gene g in experiment t, while Bfg represents the ChIP log-ratio for transcription factor f and the promoter region of gene g. The intercept F0t represents a baseline expression level, while the regression coefficients Fft can be interpreted as inferred transcription factor activities. Starting with the full set of 113 transcription factors, we used backward selection to eliminate uninformative transcription factors from our model: First, for each microarray experiment a P-value corresponding to each regression coefficient was determined, based on an F-test [26]. The transcription factors were then sorted based on the smallest P-value among all 751 experiments. In an iterative procedure, the transcription factor with the most insignificant P-value was removed until all factors were significant at a P-value of 0.005/751. Since this analysis in itself is novel and useful, we have made an online ChIP regression tool available at http://bussemaker.bio.columbia.edu/tools/.
Gene-TF coupling factor
For each pair-wise combination of a gene g (represented by its mRNA expression profile Egt) and a transcription factor f (represented by the inferred activity profile Fft), a regulatory coupling factor was calculated, equal to the Pearson correlation between Egt and Fft:
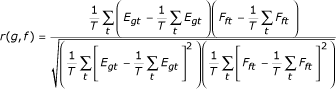
For each value of r, an associated P-value was computed by performing a t-test on t = r[(G-2)/(1-r2)]1/2. To account for the parallel testing of many TF-target pairs, but at the same time avoid the overly conservative Bonferroni correction, we set a threshold for t by requiring a false discovery rate of 5% [27]. The end result of this procedure is a list of genes that are significantly coupled to a transcription factor. Strictly speaking, to avoid circularity, the coupling of each gene should be evaluated based on a TFAP derived from expression data for all but that gene. However, as the TFAP is derived from the expression profile of all genes bound by the TF, the effect of leaving out one gene is relatively insignificant in practice. Moreover, repeating this procedure for every gene in the genome would be computationally unfeasible.
Enrichment for gene ontology categories
Based on the regulatory coupling analysis described above, the genes bound a given transcription factor (B+) were sorted in two classes, B+/C+ (bound and coupled) and B+/C- (bound but not coupled). These two sets were used as input for further analysis. The cumulative hypergeometric distribution was used to determine whether a set of genes is enriched for one or more Gene Ontology categories [20]. The Bonferroni correction was applied to all P-values to deal with the parallel testing of GO categories. The organism-independent ontology and the gene-association table (version May 2003) for S. cerevisiae were downloaded from http://www.geneontology.org.
Response to transcription factor deletion
Expression data for mutant vs. wild-type comparison for the transcription factors Dig1, Gcn4, Hir2, Mbp1, Swi4, Swi5, and Yap1 were obtained from Hughes et al. [21]. To test whether a given subset of genes responded to TF deletion, a sample t-test was performed, comparing the average expression log-ratio in the subset with the genome-wide distribution of expression changes. To guarantee that this analysis was fair, the respective TF deletion experiments were excluded from the library used to calculate the coupling factors that define the C+ and C- groups.
Enrichment for cell cycle DNA motifs
Four different DNA motifs found as top-scoring motifs by REDUCE and also reported in Spellman et al. were tested for over-representation in the set of B+/C+ and B+/C- genes, respectively, for the 7 cell-cycle related transcription factors within the set of 37 factors analyzed by us [16,28]. These motifs are: ACGCGT (MCB), CGCGAAA (SCB), AACCAGC (Swi5p) and GTAAACA (SFF). Motifs were counted in non-coding regions up to 600 bp upstream from the ORF start position, and expected counts were based on upstream regions of all genes. No overlapping matches were counted. The cumulative binomial distribution was used to assign a P-value to the enrichment for these motifs.
List of abbreviations
TF: transcription factor
ChIP: chromatin immunoprecipitation
DamID: DNA adenine methyltransferase identification
TFAP: transcription factor activity profile
GO: gene ontology.
Authors' contributions
FG and HJB both contributed to the development of the algorithm and the analysis and presentation of the results. BCF compiled the library of expression data used and contributed tools for functional annotation enrichment analysis. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Marcel van Batenburg and Crispin Roven for their assistance and helpful suggestions. We are also grateful to Frank Holstege, Bas van Steensel, and Kevin White for valuable comments and a critical reading of the manuscript. F.G. was partially funded by the Netherlands Organization for Scientific Research (NWO) and the Human Frontier Science Programme (HFSP). B.C.F. and H.J.B. were partially funded by the National Institutes of Health.
References
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. [Abstract] [Google Scholar]
- Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet. 1999;21:33–37. 10.1038/4462. [Abstract] [CrossRef] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. 10.1038/35054095. [Abstract] [CrossRef] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. 10.1126/science.1075090. [Abstract] [CrossRef] [Google Scholar]
- van Steensel B, Delrow J, Henikoff S. Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet. 2001;27:304–308. 10.1038/85871. [Abstract] [CrossRef] [Google Scholar]
- Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JC, Trent JM, Staudt LM, Hudson J., Jr., Boguski MS, Lashkari D, Shalon D, Botstein D, Brown PO. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. 10.1126/science.283.5398.83. [Abstract] [CrossRef] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. 10.1126/science.290.5500.2306. [Abstract] [CrossRef] [Google Scholar]
- van Steensel B, Delrow J, Bussemaker HJ. Genomewide analysis of Drosophila GAGA factor target genes reveals context-dependent DNA binding. Proc Natl Acad Sci U S A. 2003;100:2580–2585. 10.1073/pnas.0438000100. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Sun LV, Chen L, Greil F, Negre N, Li TR, Cavalli G, Zhao H, Van Steensel B, White KP. Protein-DNA interaction mapping using genomic tiling path microarrays in Drosophila. Proc Natl Acad Sci U S A. 2003;100:9428–9433. 10.1073/pnas.1533393100. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Wyrick JJ, Holstege FC, Jennings EG, Causton HC, Shore D, Grunstein M, Lander ES, Young RA. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402:418–421. 10.1038/46567. [Abstract] [CrossRef] [Google Scholar]
- Futcher B. Transcriptional regulatory networks and the yeast cell cycle. Curr Opin Cell Biol. 2002;14:676–683. 10.1016/S0955-0674(02)00391-5. [Abstract] [CrossRef] [Google Scholar]
- Banerjee N, Zhang MQ. Functional genomics as applied to mapping transcription regulatory networks. Curr Opin Microbiol. 2002;5:313–317. 10.1016/S1369-5274(02)00322-3. [Abstract] [CrossRef] [Google Scholar]
- Ihmels J, Friedlander G, Bergmann S, Sarig O, Ziv Y, Barkai N. Revealing modular organization in the yeast transcriptional network. Nat Genet. 2002;31:370–377. [Abstract] [Google Scholar]
- Bar-Joseph Z, Gerber GK, Lee TI, Rinaldi NJ, Yoo JY, Robert F, Gordon DB, Fraenkel E, Jaakkola TS, Young RA, Gifford DK. Computational discovery of gene modules and regulatory networks. Nat Biotechnol. 2003;21:1337–1342. 10.1038/nbt890. [Abstract] [CrossRef] [Google Scholar]
- Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet. 2001;29:153–159. 10.1038/ng724. [Abstract] [CrossRef] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat Genet. 2001;27:167–171. 10.1038/84792. [Abstract] [CrossRef] [Google Scholar]
- Wang W, Cherry JM, Botstein D, Li H. A systematic approach to reconstructing transcription networks in Saccharomycescerevisiae. Proc Natl Acad Sci U S A. 2002;99:16893–16898. 10.1073/pnas.252638199. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Keles S, van der Laan M, Eisen MB. Identification of regulatory elements using a feature selection method. Bioinformatics. 2002;18:1167–1175. 10.1093/bioinformatics/18.9.1167. [Abstract] [CrossRef] [Google Scholar]
- Conlon EM, Liu XS, Lieb JD, Liu JS. Integrating regulatory motif discovery and genome-wide expression analysis. Proc Natl Acad Sci U S A. 2003;100:3339–3344. 10.1073/pnas.0630591100. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 2000;25:25–29. 10.1038/75556. [Abstract] [CrossRef] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. 10.1016/S0092-8674(00)00015-5. [Abstract] [CrossRef] [Google Scholar]
- Simon I, Barnett J, Hannett N, Harbison CT, Rinaldi NJ, Volkert TL, Wyrick JJ, Zeitlinger J, Gifford DK, Jaakkola TS, Young RA. Serial regulation of transcriptional regulators in the yeast cell cycle. Cell. 2001;106:697–708. 10.1016/S0092-8674(01)00494-9. [Abstract] [CrossRef] [Google Scholar]
- Li TR, White KP. Tissue-specific gene expression and ecdysone-regulated genomic networks in Drosophila. Dev Cell. 2003;5:59–72. 10.1016/S1534-5807(03)00192-8. [Abstract] [CrossRef] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. 10.1038/ng1165. [Abstract] [CrossRef] [Google Scholar]
- Zhu Z, Pilpel Y, Church GM. Computational identification of transcription factor binding sites via a transcription-factor-centric clustering (TFCC) algorithm. J Mol Biol. 2002;318:71–81. 10.1016/S0022-2836(02)00026-8. [Abstract] [CrossRef] [Google Scholar]
- Jobson JD. Springer texts in statistics. New York, Springer-Verlag; 1991. Applied multivariate data analysis; p. 2 v.. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B. 1995;57:289–300. [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. [Europe PMC free article] [Abstract] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. [Europe PMC free article] [Abstract] [Google Scholar]
- Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett HA, He YD, Dai H, Walker WL, Hughes TR, Tyers M, Boone C, Friend SH. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000;287:873–880. 10.1126/science.287.5454.873. [Abstract] [CrossRef] [Google Scholar]
Articles from BMC Bioinformatics are provided here courtesy of BMC
Full text links
Read article at publisher's site: https://doi.org/10.1186/1471-2105-5-31
Read article for free, from open access legal sources, via Unpaywall:
https://bmcbioinformatics.biomedcentral.com/counter/pdf/10.1186/1471-2105-5-31

Citations & impact
Impact metrics
Citations of article over time
Article citations
Identifying genetic regulatory variants that affect transcription factor activity.
Cell Genom, 3(9):100382, 18 Aug 2023
Cited by: 1 article | PMID: 37719147 | PMCID: PMC10504674
PathwayMultiomics: An R Package for Efficient Integrative Analysis of Multi-Omics Datasets With Matched or Un-matched Samples.
Front Genet, 12:783713, 22 Dec 2021
Cited by: 8 articles | PMID: 35003218 | PMCID: PMC8729182
Filtering of Data-Driven Gene Regulatory Networks Using Drosophila melanogaster as a Case Study.
Front Genet, 12:649764, 28 Jul 2021
Cited by: 3 articles | PMID: 34394179 | PMCID: PMC8355599
Inferring TF activities and activity regulators from gene expression data with constraints from TF perturbation data.
Bioinformatics, 37(9):1234-1245, 01 Jun 2021
Cited by: 11 articles | PMID: 33135076 | PMCID: PMC8189679
Transcriptional network modulated by the prognostic signature transcription factors and their long noncoding RNA partners in primary prostate cancer.
EBioMedicine, 63:103150, 03 Dec 2020
Cited by: 8 articles | PMID: 33279858 | PMCID: PMC7718452
Go to all (141) article citations
Data
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
Identification of transcription factor targets by phenotypic activation and microarray expression profiling in yeast.
Methods Mol Biol, 548:19-35, 01 Jan 2009
Cited by: 4 articles | PMID: 19521817
Transcriptional regulatory networks in Saccharomyces cerevisiae.
Science, 298(5594):799-804, 01 Oct 2002
Cited by: 1596 articles | PMID: 12399584
Genome-wide prediction of transcriptional regulatory elements of human promoters using gene expression and promoter analysis data.
BMC Bioinformatics, 7:330, 04 Jul 2006
Cited by: 20 articles | PMID: 16817975 | PMCID: PMC1586028
Transcriptional networks: reverse-engineering gene regulation on a global scale.
Curr Opin Microbiol, 7(6):638-646, 01 Dec 2004
Cited by: 39 articles | PMID: 15556037
Review


