Abstract
Free full text
Modeling RNA-binding protein specificity in vivo by precisely registering protein-RNA crosslink sites
Summary
RNA-binding proteins (RBPs) regulate post-transcriptional gene expression by recognizing short and degenerate sequence motifs in their target transcripts, but precisely defining their binding specificity remains challenging. Crosslinking and immunoprecipitation (CLIP) allows for mapping of the exact protein-RNA crosslink sites, which frequently reside at specific positions in RBP motifs, at single-nucleotide resolution. Here, we have developed a computational method, named mCross, to jointly model RBP binding specificity while precisely registering the crosslinking position in motif sites. We applied mCross to 112 RBPs using ENCODE eCLIP data and validated the reliability of the discovered motifs by genome-wide analysis of allelic binding sites. Our analyses revealed that the prototypical SR protein SRSF1 recognizes clusters of GGA half sites in addition to its canonical GGAGGA motif. Therefore, SRSF1 regulates splicing of a much larger repertoire of transcripts than previously appreciated, including HNRNPD and HNRNPDL, which are involved in multivalent protein assemblies and phase separation.
Introduction
RNA-binding proteins (RBPs) are central for post-transcriptional regulation of gene expression by interacting with specific sequence or structural elements embedded in their target transcripts (Licatalosi and Darnell, 2010). Precise characterization of RBP binding specificity is crucial for identifying functional protein-RNA interaction sites regulating gene expression and for understanding how such interactions are affected by genetic variation across individuals in a population, particularly in the context of human disease (Lunde et al., 2007).
Multiple approaches have been used to determine RBP binding sites and define RBP binding specificity in vitro and in vivo (Jankowsky and Harris, 2015). UV cross-linking and immunoprecipitation (CLIP) of protein-RNA complexes, followed by high-throughput sequencing of isolated RNA fragments (HITS-CLIP or CLIP-seq), is a biochemical assay to map in vivo protein-RNA interactions on a genome-wide scale (Licatalosi et al., 2008; Ule et al., 2005; Ule et al., 2003). Since its initial development, CLIP and multiple variant protocols have been applied to an expanding list of RBPs in various species and cellular contexts (Darnell, 2010; Licatalosi and Darnell, 2010). In particular, a modified version of CLIP, named enhanced CLIP (eCLIP), was adopted by the Encyclopedia of DNA Elements (ENCODE) consortium to map the binding sites of over 100 RBPs in two human cell lines, HepG2 and K562, making it the largest CLIP dataset generated thus far (Van Nostrand et al., 2017; Van Nostrand et al., 2016).
Both in vitro binding assays (such as RNAcompete (Ray et al., 2009; Ray et al., 2013)) and CLIP generate a list of sequences expected to be bound by an RBP. A common pattern shared by these sequences, or motif, needs to be inferred de novo by statistical modeling to define the sequence specificity of the RBP and predict individual binding sites. A similar task is present for studies of DNA-binding proteins, which were historically the initial focus of genomic analysis using large-scale datasets. Therefore, some of the current methods used for de novo RBP motif discovery (e.g., MEME and HOMER) were originally developed for analysis of DNA-binding proteins (Bailey and Elkan, 1994; Heinz et al., 2010). However, there exist important differences between DNA-binding proteins and RBPs. As compared to DNA-binding proteins, most RBPs recognize very short (~3–7 nt) and degenerate sequence motifs, which generally have limited information content (Chen and Manley, 2009; Jankowsky and Harris, 2015; Lunde et al., 2007; Singh and Valcarcel, 2005). For example, the high-affinity binding motif of the neuron-specific splicing factor Nova is the tetramer YCAY (Y=C/U) (Jensen et al., 2000). Due to the apparently lower specificity of RBPs, the performance of the current computational tools for de novo motif discovery varies when applied to RBPs. Consequently, despite the availability of CLIP or high-throughput in vitro binding data, the specificity of many RBPs remains to be defined. This challenge is reflected in situations in which distinct motifs have been reported for the same RBPs from different datasets (e.g., for FMRP (Ascano et al., 2012; Darnell et al., 2005; Darnell et al., 2001; Darnell et al., 2011)). In addition, multiple RBPs were reported to have similar motifs and yet they have very distinct binding maps in the transcriptome (e.g., for TIA1, hnRNP C and other RBPs recognizing U-rich or AU-rich elements (Konig et al., 2010; Wang et al., 2010)).
The degeneracy of RBP binding motifs argues for the importance of mapping RBP binding sites with high resolution to improve accuracy of motif discovery. Previously, we developed computational approaches to map the extract protein-RNA crosslink sites through analysis of crosslink-induced mutation sites (CIMS) and truncation sites (CITS) using CLIP data (Weyn-Vanhentenryck et al., 2014; Zhang and Darnell, 2011). CIMS and CITS are signatures of protein-RNA crosslinking introduced when the covalently linked amino acid-RNA adducts interfere with reverse transcription. Importantly, CIMS and CITS provide a means of mapping protein-RNA interactions at single-nucleotide resolution. Furthermore, our previous analysis has revealed that UV crosslinking frequently occurs at specific positions in the RBP binding motifs, most likely reflecting the critical RNA residues for direct protein-RNA contacts (e.g., G2 and G6 in UGCAUG that is recognized by RBFOX) (Moore et al., 2014; Weyn-Vanhentenryck et al., 2014).
Here we report that these crosslink sites can be used to precisely register RBP binding sites, at single-nucleotide resolution, to improve the accuracy of de novo RBP motif discovery. We demonstrate the effectiveness of this strategy by developing a statistical model and algorithm named mCross and applying it to 112 RBPs using ENCODE eCLIP data. The reliability of the resulting motifs defined by mCross was validated by analysis of allelic protein-RNA interaction sites caused by heterozygous SNPs on a genome-wide scale. Based on motifs defined by mCross, we unexpectedly found that SRSF1, a prototypical SR protein, predominantly recognizes clusters of GGA half sites, instead of just recognizing its canonical GGAGGA motif, which allows it to regulate splicing of a much larger repertoire of transcripts than previously appreciated. Among the previously uncharacterized targets are the alternative exons within HNRNPD and HNRNPDL, which were previously demonstrated to modulate multivalent hnRNP assemblies and phase separation (Gueroussov et al., 2017). Finally, we have developed a searchable, interactive web interface (http://zhanglab.c2b2.columbia.edu/index.php/MCross) to facilitate use of this resource by the research community.
Results
Joint modeling of RBP binding specificity and precise crosslinking positions
Most of the de novo motif discovery tools currently available use a position-specific weight matrix (PWM) (Stormo, 2000) to characterize the specificity of DNA- or RNA-binding proteins (note that the consensus can be viewed as a special case of a PWM). mCross takes advantage of the precise protein-RNA crosslink sites inferred from CIMS and CITS analysis and the observation that crosslinking frequently occurs at specific positions within the motif (Figure 1A,,B).B). Therefore, mCross augments the standard PWM model by jointly modeling RBP sequence specificity and the precise protein-RNA crosslink positions so that it can register motif sites in longer input sequences at single-nucleotide resolution to dramatically limit the search space (Figure 1C). Optimal model parameters are determined by maximizing the likelihood function (see STAR Methods).

Related to Figures S1 and S2.
(A) Identification of reproducible protein-RNA crosslink sites by analysis of crosslinking-induced mutation sites (CIMS) and truncation sites (CITS).
(B) Enrichment of top 7-mers in sequences around Rbfox crosslink sites. Crosslink sites were identified by CIMS analysis of deletions and the frequency of each 7-mer starting at different positions relative to the crosslink site is shown in the heatmap. Representative 7-mers showing the highest position-specific enrichment are indicated and the corresponding crosslinked nucleotide is highlighted in red.
(C) The likelihood function that jointly models RBP sequence specificity and crosslinking positions is shown at the top. The Rbfox binding motif and the crosslinking probability at each position of the motif discovered de novo by mCross are shown at the bottom.
(D) Additional examples of RBP motifs and crosslink sites as discovered de novo by mCross.
For initial assessment on the reliability of mCross, we applied it to several tissue-specific RBPs, including Rbfox (Weyn-Vanhentenryck et al., 2014), Nova (Zhang et al., 2010), Ptbp2 (Licatalosi et al., 2012), Mbnl2 (Charizanis et al., 2012), and Lin28a (Cho et al., 2012), which have variable binding specificities that have been previously well characterized by CLIP and other experimental approaches. mCross simultaneously recovered a well-defined motif for every RBP and determined the predominant crosslink sites within the motif (Figure 1C,,D).D). For example, Rbfox binds the (U)GCAUG element with a certain level of degeneracy at the first position, U1, and the predominant crosslink sites are G2 and G6. Ptbp2 binds to UCUCU-like elements with predominant crosslink sites at the cytosines. Importantly, all motifs defined for the same RBP, as discovered by mCross, are highly similar to each other, minimizing the ambiguity in determining bona fide specificity (Figure S1). These results also confirmed that photocrosslinking can occur at different nucleotides, although uridine-bias is assumed based on previous literature.
To further evaluate the effectiveness of mCross, we applied it to Argonaute (Ago) CLIP data derived from mouse brain (Chi et al., 2009) to recover microRNA (miRNA) binding sites that are reverse complementary to seed sequences. Since Ago mRNA CLIP tags could capture binding sites of all miRNAs expressed in the brain, the data represent a mixture of multiple motifs. mCross successfully identified motifs that can be grouped into 10 clusters, including canonical seed matches of six miRNAs that are abundantly expressed in the brain and the miR-124 seed matches with a bulge (Figure S2) (Chi et al., 2012). In general, Ago preferentially crosslinks to target mRNA transcripts at positions flanking seed matches, but in some cases it appears that uridines inside the seed matches are also prone to crosslinking. This example demonstrates that mCross is capable of deconvoluting different modes of binding in more complex datasets.
Defining motifs of 112 unique RBPs using ENCODE eCLIP data
Having demonstrated the promise of mCross, we extended our analysis to eCLIP data from ENCODE (Van Nostrand et al., 2017). These include 70 RBPs in HepG2 cells, 89 RBPs in K562 cells, and 1 RBP in adrenal gland, with each RBP assayed in two replicate experiments (in total, 160 experiments×2=320 independent CLIP libraries, representing 112 unique RBPs; as of Dec 30, 2016). The replicates allow for evaluation of reproducibility (see below). All CLIP data were processed using our established CTK package (Shah et al., 2017) to call CLIP tag cluster peaks and infer potential crosslink sites using CIMS and CITS analysis (Figure S3A–C and Table S1).
Since the majority of RBPs assayed by eCLIP were previously poorly characterized, we developed quantitative metrics to evaluate if an RBP likely has robust binding specificity. First, we estimated the number of 7-mers that are asymmetrically enriched in CLIP tag clusters as compared to the number of 7-mers that are depleted. Second, we developed a ‘disconcordance’ score (D-score) to measure whether the top 7-mers are consistently enriched in the two biological replicates, with a low D-score indicating high concordance between the replicates (see STAR Methods). For example, RBFOX2 showed a very low D-score between the two replicates (D<0.00067), and most of the significantly enriched 7-mers contained the (U)GCAUG motif that is known to bind the protein (Figure 2A).

Related to Figure S3.
(A) Illustration of top 7-mer disconcordance and asymmetric enrichment using RBFOX2 eCLIP in HepG2 cells for an example. A z-score was calculated for each 7-mer based on its enrichment in CLIP tag cluster peaks for each replicate. 7-mers asymmetrically enriched in peaks are indicated using the blue box and 7-mers containing (U)GCAUG are highlighted using red circles.
(B) RBPs are ranked based on the top 7-mer disconcordance (D)-scores between peaks of the two replicates, between peaks and CITS, and between peaks and CIMS (deletions, substitutions and insertions analyzed separately).
(C) Number of asymmetrically enriched top 7-mers for each RBP (q<0.05), shown in the same order as in (B).
Top 7-mers in 88/160 (55%) CLIP experiments showed D-scores <0.05 (indicating that the top 20 7-mers in replicate A have an average ranking of 0.05×47/2=~400 in replicate B and vice versa; Figure 2B). For RBPs with low D-scores between the two replicates in the same cell lines, they also have low D-scores when CLIP data from different cell lines were compared, indicating the same binding specificity in different cell types (Figure S3D). In addition, RBPs with low D-scores in general have a larger number of significantly enriched 7-mers (Spearman ρ=−0.7, p<4.7e-25; Figure 2C). These observations together suggest that RBPs with low D-scores are more likely to have robust and reproducible binding specificity. While some RBPs with large D-scores between replicates could be due to technical issues, they might also tend to lack recognizable sequence specificity. In line with this notion, we found that a significantly higher portion of RBPs with D<0.05 have annotated RNA-binding domains (RBDs)(Cook et al., 2011), as compared to RBPs with D>0.05 (51/88=58% vs. 24/72=35%; p=0.0042; Fisher’s exact test). The enrichment is also clear for individual types of RBDs, such as RNA-recognition motifs (RRMs) (36/88=41% vs. 17/72=24%; p=0.028; Fisher’s exact test) or K homology (KH) domains (14/88=16% vs. 1/72=1.4%; p=0.0018; Fisher’s exact test). These observations presumably reflect the fact that these RBDs are well known to recognize specific sequences.
We used the D-score metric to compare top 7-mers enriched in CLIP tag cluster peaks and those enriched in CITS or CIMS derived from different types of mutations to assess reliability of inferred crosslink sites. Among the 88 CLIP experiments with D<0.05 between replicates, 38 showed consistent 7-mer enrichment (D<0.05) compared to CITS, while few showed consistent 7-mer enrichment in CIMS. Based on these and other observations, we concluded that CIMS does not appear to provide reliable inference of crosslink sites in this dataset, and we focused on crosslink sites inferred by CITS for de novo motif discovery using mCross.
We thus applied mCross to CITS identified in the 160 CLIP experiments (with two replicates combined) and were able to discover one or more motifs in 144 experiments (the other 16 experiments do not show significantly enriched 7-mers, indicating lack of binding specificity). Overall, RBPs with low D-scores between replicates tended to have more unambiguous motifs after similar ones were clustered together (Spearman rank correlation ρ=0.54, p=2.5e-12; Figure 3 and Table S2). An interactive, searchable web interface was also developed to facilitate access to this resource by the research community (http://zhanglab.c2b2.columbia.edu/index.php/MCross).

Results are shown for 38 eCLIP experiments (replicates combined) representing 27 distinct RBPs with D<0.05 between peaks of the two replicates and between peaks and CITS. Motif logos are only shown for the 27 distinct RBPs. For RBPs with multiple clusters of motifs, the representative motif with the highest likelihood score from the top cluster is shown.
Validating RBP motifs by allelic protein-RNA interactions
A major challenge for de novo motif discovery is that an algorithm typically finds multiple motifs, leaving the user to determine which one is most reliable, if any. Mutagenesis, together with measurement of binding affinity or reporter assays, is the standard approach for experimental validation of computationally identified motifs, but such validation is typically limited to a small set of selected binding sites, resulting in uncertainty in generalizability. We argued that protein-RNA interaction sites overlapping with single-nucleotide polymorphisms (SNPs) represent a large number of natural perturbation experiments. In particular, the binding affinity of the two alleles at heterozygous SNPs can be directly compared by the allelic imbalance of CLIP tags, with more CLIP tags indicating stronger binding (Figure 4A). We therefore performed allelic interaction (AI) analysis using heterozygous SNPs called from whole genome sequencing and eCLIP data in HepG2 and K562 cells (Figure S4 and Table S3; see STAR Methods for detail). In total, we identified 39,528 potential AI sites from HepG2 (an average of 565 sites per RBP) and 29,463 sites from K562 cells (an average of 331 sites per RBP; Table S4).

Related to Figures S4 and S5.
(A) Schematic of a heterozygous SNP affecting Rbfox binding and the resulting allelic imbalance in eCLIP data.
(B) The relationship between the allelic bias of CLIP tags and the motif scores of the reference and alternative alleles overlapping with SNPs using RBFOX2 and QKI eCLIP in HepG2 cells are shown for examples. In each panel, each dot represents a SNP with significant allelic imbalance in CLIP data. SNPs with more alternative allele tags are shown in red and those with more reference allele tags are shown in cyan. X-axis shows the maximum motif score for either the reference or alternative allele of all positions overlapping with the SNP, measuring the likelihood of RBP binding at least one allele. Y-axis shows the difference of the maximum motif score among all positions overlapping with the alternative allele and the maximum motif score among all positions overlapping with the reference allele, measuring the impact of the SNP on RBP binding. SNPs with consistent allelic bias in CLIP tags and motif score changes are shown in circles while inconsistent SNPs are shown in triangles.
(C) The number of consistent and inconsistent AI SNP sites for each RBP (top). For this analysis, the top PWM was used as a representative for each RBP. The excess of consistent over inconsistent AI sites is shown at the bottom and RBPs are color-coded based on the extent of excess using a Binomial test.
(D) Comparison of mCross and other de novo motif discovery programs using eCLIP data and AI analysis. The number of AI-consistent and AI-inconsistent PWMs (one PWM per RBP) discovered by each program using different significance thresholds is shown. For each RBP, the top PWM discovered by each method was used for comparison.
(E) Among the AI-consistent PWMs discovered by each program at p<0.05, the number of RBPs showing low (D≤0.05) or high (D>0.05) D-scores is shown.
If the allelic imbalance detected in CLIP data is indeed due to differential binding of the implicated RBP to the reference or alternative alleles, and the motif model accurately characterizes the binding specificity of the RBP, one would expect that the allele with more CLIP tags would have a higher motif score, and the other allele (with fewer CLIP tags) would have a lower motif score. We denote these AI sites “consistent” sites (and the opposite case as “inconsistent” AI sites). For example, for the AI sites of RBFOX2 and QKI with more CLIP tags supporting the alternative allele (“red points”), the motif score is in general higher for the alternative allele, while for the AI sites with fewer CLIP tags supporting the alternative allele (“cyan points”), the motif score is in general higher in the reference allele (Figure 4B). As expected, the trend is clear only for SNPs overlapping with a high-scoring motif site. For each RBP motif, we can thus obtain a subset of AI sites overlapping with high motif scores in either the reference or the alternative allele and also large motif score differences between the two alleles. The proportion of consistent AI sites in this subset can be used as a measure of motif model accuracy.
We initially used a representative PWM (the first PWM discovered by mCross) for each RBP to compare with AI sites to avoid any bias, as selection of the PWM is independent of AI site analysis. For the majority of RBPs, consistent AI sites are in excess as compared to inconsistent AI sites (90 vs. 39 RBPs; Figure 4C). Using a more stringent threshold (p<0.05; Binomial test), 26 RBPs have significantly more consistent AI sites compared with inconsistent AI sites (denoted AI-consistent PWMs) and only 2 RBPs show the opposite pattern (Figure 4C). For the remaining RBPs, which individually have an insufficient number of AI sites for statistical analysis, the overall proportion of consistent AI sites is also significantly higher than 0.5 when they were analyzed in aggregate (p<0.004; Binomial test). Therefore, the concordance between the allelic imbalance of CLIP tags and changes in motif scores provides unbiased validation that the motifs defined by mCross reliably reflect RBP binding specificity. Furthermore, these AI sites also provide a list of genetic variants in the human populations that directly affect protein-RNA interactions with potential impact on downstream post-transcriptional gene expression regulation and individual phenotypes.
Comparison of mCross with other motif discovery methods by allelic interaction sites
We next used AI site analysis to evaluate PWMs derived from other methods to provide an unbiased comparison of these methods to mCross. First, we compared mCross with several programs widely used for de novo motif discovery, including MEME (Bailey and Elkan, 1994), DREME (Bailey, 2011), HOMER (Heinz et al., 2010), and Zagros (Bahrami-Samani et al., 2015). The comparison of mCross with Zagros is particularly worth noting because Zagros also uses the proximity of diagnostic crosslinking events for motif discovery, although mCross differs from Zagros in several critical aspects (see Discussion). To simplify comparison, we used the top motif found by each program for every RBP from the same input sequence data. mCross consistently outperformed the other programs in terms of the number and proportion of AI-consistent PWMs defined using different p-value thresholds (Figure 4D). For example, among the 159 PWMs derived from HepG2 and K562 data by MEME, 24 were AI-consistent PWMs and 5 were AI-inconsistent PWMs (p<0.05; Binomial test) as compared to 26 AI-consistent PWMs and 2 AI-inconsistent PWMs derived by mCross using the same criteria. If AI-consistent PWMs are more reliable, we expect them to have low D-scores between CLIP replicates (Figure 4E). Indeed, for the 14 RBPs with AI-consistent PWMs identified by both mCross and MEME, all have D<0.05. Notably, only 1 of 12 RBPs with AI-consistent PWMs exclusively identified by mCross have D>0.05, while 4 of 10 RBPs with AI-consistent PWMs exclusively identified by MEME have D>0.05 (Figure S5A). At a threshold p<0.05, Zagros identified 25 PWMs, including 24 that are AI-consistent, which is close to mCross. However, among the 14 AI-consistent PWMs identified exclusively by Zagros, 7 (50%) have D>0.05, as compared to 1 of 16 (6%) PWMs identified exclusively by mCross but not Zagros (Figure S5B). In addition, with more stringent p-value thresholds, mCross identified substantially more AI-consistent PWMs than Zagros (11 vs. 6 at p<0.005; Figure 4D). Taken together, motifs derived by mCross showed better concordance with AI sites than those derived by other compared programs, suggesting that mCross is able to characterize RBP binding specificity more accurately.
We also compared 18 RBPs with both eCLIP and RNAcompete data (Ray et al., 2013). Among them, 6 RBPs have AI-consistent PWMs derived from RNAcompete, and 10 RBPs have AI-consistent PWMs derived by mCross. For the four RBPs with AI-consistent PWMs only identified by mCross (HNRNPA1, TARDBP, TIA1, and U2AF2), all have well characterized binding specificity.
Given the effectiveness of AI sites for evaluating different PWMs for the same RBPs, we selected the optimal motifs for every RBP based on their consistency with AI sites (Table S5). This analysis allowed us to obtain a final list of 16 RBPs in HepG2 and 13 RBPs in K562 with AI-consistent PWMs (FDR<0.1; Figure 5) and a subset of AI sites filtered by these PWMs that most likely directly affect protein-RNA interactions (Table S4).

Only RBPs with AI-consistent PWMs at FDR<0.1 are shown.
SRSF1 recognizes clusters of GGA half sites to activate exon inclusion
Overall, for well characterized RBPs, the motifs defined by mCross agree with the previously defined motifs (Figures 3 and and5).5). However, there are also interesting exceptions. For example, we found a (U)GAU motif for LIN28, which is distinct from its previously characterized GGAG motif (Cho et al., 2012; Wilbert et al., 2012). The functional significance of this (U)GAU motif was described in our recent study (Ustianenko et al., 2018). Here we focus on another example of a distinct motif identified by mCross, specifically for the RBP SRSF1, and we discuss the importance of this motif for splicing regulation.
SRSF1 is the founding member of the SR protein family, which is important for regulation of both constitutive and alternative splicing (Long and Caceres, 2009). mCross identified a UGGA motif for SRSF1 with predominant crosslinking in the U1 position. The GGA motif represents a half site of the previously identified SRSF1-binding consensus GGAGGA using SELEX, RNAcompete, and CLIP data (Ray et al., 2013; Sanford et al., 2009; Tacke and Manley, 1995) (Figure 6A). The first uridine of the UGGA motif likely reflects crosslinking bias, as SNPs at this position do not affect binding, while the other three positions are biologically important (Figure S6A). Interestingly, a previous structural study suggested that the second RRM of SRSF1 directly contacts a GGA half site (Figure 6B) (Clery et al., 2013), which agrees well with this distinct motif discovered by mCross.

Related to Figure S6.
(A) SRSF1 binding motif determined by mCross (GGA) and by RNAcompete (GGAGGA).
(B) The NMR structure of SRSF1 RRM2-RNA complex (PDB accession: 2m8d). The GGA half site directly contacting the RRM is highlighted in green.
(C) AI analysis using bipartite GGA-N[0,2]-GGA motif. A Binomial test was used to evaluate whether the excess of consistent AI sites over inconsistent AI sites is significant (* p<0.05).
(D) Overlap between predicted GGA clusters and SRSF1 eCLIP tag clusters at different ranks of motif scores. GGA clusters without overlapping with GGAGGA and conserved GGAGGA sites ranked by BLS are shown for comparison. Only motif sites in the CDS region were used for this analysis.
(E, F) RNA map showing predicted GGA clusters enriched in cassette exons with SRSF1-dependent inclusion, but depleted in alternative exons with SRSF1-dependent skipping. Results were obtained for all GGA clusters (E) or GGA clusters without overlapping with GGAGGA (F).
(G, H) Classification of SRSF1-activated cassette exons using predicted GGA clusters. Results based on conserved GGAGGA sites and CLIP tag clusters are shown for comparison. The sensitivity (G) or positive prediction value (H) is shown at varying stringencies for each method.
(I) Among the SRSF1-dependent cassette exons harboring predicted GGA clusters, the number of exons with or without overlapping canonical GGAGGA motif is shown.
(J) Predicted GGA clusters are enriched in exons with high inclusion level. Cassette exons are binned based on exon inclusion level in HepG2 cells. For each bin, the percentage of exons with predicted GGA clusters above varying thresholds is shown.
(K) Similar to (J), but using predicted GGA clusters excluding those overlapping with the GGAGGA motif.
(L) Among the highly included cassette exons (Ψ ≥0.9) harboring predicted GGA clusters, the number of exons with or without overlapping GGAGGA motif is shown.
Since the GGA motif alone has very limited information content, we reasoned that sufficient targeting specificity for SRSF1 has to be achieved by binding to a cluster of GGA half sites in proximity to one another. Mechanistically, GGA clusters can be bound by multimerization of SRSF1 proteins with each RRM contacting one GGA motif site (Liu et al., 1998). To test this hypothesis, we first searched for bipartite GGA-Nx-GGA motifs with a spacer. Indeed, we found an excess of consistent AI sites overlapping GGAN[1–2]GGA, similar to the pattern found for the canonical GGAGGA motif (Figure 6C). We therefore predicted GGA clusters using mCarts, which integrates the number of GGA sites, their spacing, cross-species conservation, and accessibility as determined by predicted RNA-secondary structures (Weyn-Vanhentenryck and Zhang, 2016; Zhang et al., 2013) (Figure S6B). GGA clusters predicted using the models trained by HepG2 and K562 CLIP data are highly similar to each other qualitatively and quantitatively (Figure S6C,D). Therefore, GGA clusters trained on HepG2 CLIP data were used for detailed analysis described in this study.
The predicted GGA clusters with higher motif scores in general have a larger overlap with CLIP tag clusters (up to over 40%, as compared to 18% overlap observed from individual GGA elements; Figure 6D). This is true after excluding clusters containing GGAGGA, suggesting SRSF1 binds to GGA clusters without requiring GGAGGA on a genome-wide scale.
To test whether the predicted GGA clusters are sufficient to regulate alternative splicing, we identified cassette exons showing altered splicing upon SRSF1 knockdown in HepG2 and K562 cells, using RNA-seq data generated by ENCODE (Van Nostrand et al., 2017). As a positive control, we first generated an RNA map that predicts the impact of SRSF1 binding position on splicing using CLIP tags and the canonical GGAGGA motif sites. As expected, substantial enrichment of SRSF1 binding was observed in alternative exons with SRSF1-dependent inclusion, and depletion of SRSF1 binding was observed in cassette exons with SRSF1-dependent skipping (Figure S6E,F). This map is consistent with the known role of SRSF1 in activating exon inclusion by binding to exonic splicing enhancers (ESEs). Importantly, the same pattern was obtained using predicted GGA clusters, even after excluding GGA clusters overlapping with GGAGGA (Figure 6E,,F).F). These results suggest that the predicted GGA clusters without GGAGGA are functional in activating exon inclusion.
We next evaluated how well GGA cluster motif scores predict individual SRSF1 target exons. To this end, we scored every cassette exon by the strongest GGA cluster in the exon. Exons ranked by conserved GGAGGA using branch length score (BLS) (Zhang et al., 2008) and CLIP tag cluster scores were used for comparison. Among exons with SRSF1-dependent inclusion in both HepG2 and K562 cells (change in percent spliced in (ΔΨ)>0.1 and false discovery rate (FDR)<0.05), 54% (200/373) have predicted GGA clusters, as compared to 21% among all cassette exons. We ranked exons by their scores and calculated the sensitivity and positive prediction value (PPV) of predicting SRSF1-activated exons captured at each rank. This allowed us to compare the performance of GGA clusters and the canonical GGAGGA motif in defining SRSF1-dependent splicing regulation. We found that the GGA clusters are more predictive than conserved GGAGGA motif sites, as reflected in an increase in both sensitivity and PPV (Figure 6G,,H).H). Importantly, the performance of the GGA clusters is similar to, if not higher than, that of CLIP tag cluster scores and has overall more scored exons, indicating that the GGA clusters are both reliable and complementary to the CLIP data. Excluding GGA clusters overlapping with GGAGGA remains predictive of SRSF1-dependent exons, despite a minor reduction in performance (Figure 6G,,H).H). In fact, the majority of SRSF1-dependent exons harboring GGA clusters (137/200=69%) do not overlap with the canonical GGAGGA motif (Figure 6I).
We also reasoned that SRSF1-dependent exons identified through knockdown experiments might underestimate the contribution of SRSF1 in splicing regulation due to compensation by other SR proteins or other mechanisms. To address this issue, we examined whether GGA clusters are predictive of exon inclusion level. Indeed, exons with high inclusion are enriched in predicted GGA clusters, even after exclusion of GGAGGA, which is consistent with their role as ESEs. In HepG2 cells, 30.2% of cassette exons with inclusion level Ψ≥0.9 have predicted GGA clusters, as compared to 8.8% for cassette exons with Ψ ≤0.1, suggesting a conservative estimate of over 20% cassette exons with high inclusion as SRSF1 targets (Figure 6J,,K).K). Over 80% of these GGA clusters do not overlap with GGAGGA (Figure 6L) and similar results were obtained when more stringent thresholds on the motif score were used. Altogether, our analysis suggests that SRSF1 has a much larger repertoire of transcripts that it can recognize to regulate their splicing.
SRSF1 regulates HNRNPD and HNRNPDL alternative splicing involved in phase separation
Among SRSF1-dependent alternative exons, we found that SRSF1 strongly regulates cassette exons in both HNRNPD (exon 7; ΔΨ=0.34, FDR=1.9e-198) and HNRNPDL (exon 6; ΔΨ=0.64, FDR=1.9e-159; Figure 7A,,E)E) (these changes are in K562 cells; consistent changes were found in HepG2 cells, although somewhat smaller in magnitude). Intriguingly, in each case, the alternative exon encodes an intrinsically disordered region enriched in a glycine and tyrosine (GY) dipeptide motif, which was previously shown to mediate multivalent hnRNP assemblies with global impact on downstream splicing regulation (Gueroussov et al., 2017). In both cases, GGA clusters were predicted in the alternative exon and supported by robust CLIP tag clusters. There are two GGA clusters in HNRNPDL without any canonical GGAGGA motif sites. The GGA cluster in HNRNPD has a GGAGGA motif with four additional GGA sites separated by a variable number of nucleotides.

Related to Figure S7.
(A-D) HNRNPD.
(E-H) HNRNPDL.
(A, E) HNRNPD exon 7 (A) and HNRNPDL exon 6 (E) are bound by SRSF1 and show reduced exon inclusion upon SRSF1 depletion. RNA-seq data with and without SRSF1 knockdown and SRSF1 eCLIP data in K562 cells are shown. In each case, the cassette exon encodes an intrinsically disordered peptide enriched in glycine-tyrosine (GY) motif mediating multivalent hnRNP assemblies.
(B, F) Diagram (not to scale) of wild-type (WT) and mutant (mut) HNRNPD (B) and HNRNPDL (F) minigenes. Mutations in GGA or GGAGGA motif are indicated. For HNRNPD, three different mutants (mut-1, mut-2 and mut3) were tested.
(C, G) Splicing of WT and mut HNRNPD (C) and HNRNPDL (G) minigenes in HeLa cells, as measured by radioactive RT-PCR analysis. Representative gel images are shown on the left and average exon inclusion levels (Ψ) quantified using three independent replicates are shown on the right. Error bars represent standard deviation (SD).
(D, H) Splicing of WT and mut HNRNPD (D) and HNRNPDL (H) minigenes in HeLa cells upon depletion of SRSF1 using two different siRNAs (siSRSF1-N1 and siSRSF1-N2). siRNA against luciferase (siCt) was used as control. SRSF1 expression and minigene splicing products were measured by immunoblots (top) and radioactive RT-PCR analyses (middle), respectively. Average exon inclusion levels quantified using three independent replicates are shown with samples in the same order (bottom). Error bars represent SD.
(I) A proposed model for SRSF1 modulating multivalent hnRNP assemblies, phase separation and the downstream AS program through regulation of hnRNP alternative exons encoding GY-rich peptides.
To directly validate the importance of predicted GGA clusters for SRSF1-dependent splicing regulation, we constructed a wild type (WT) HNRNPD minigene spanning exons 6 to 8 (a partial segment of intron 7 is deleted, see STAR Methods for detail; Figure 7B). We tested splicing of the WT minigene in HeLa cells and observed nearly complete inclusion of exon 7 (average Ψ=1). HNRNPD exon 7 harbors five GGA half sites and one GGAGGA motif. To characterize the importance of each type of motif (GGA versus GGAGGA) on exon 7 splicing, we generated three additional versions of this minigene by mutating each motif type individually or simultaneously (GGA→TTA; Figure 7B). We found that mutating GGA half sites (mut-1) resulted in a much stronger loss of exon 7 inclusion (average Ψ=0.15) compared to mutating the GGAGGA motif (mut-2; average Ψ= 0.59). In addition, simultaneous mutation of both GGA and GGAGGA motifs resulted in complete loss of exon inclusion (Figure 7C). We confirmed that the regulatory effects of the tested motifs depend on SRSF1, as knockdown of SRSF1 using two independent siRNAs (siSRSF1-N1 and siSRSF1-N2) greatly reduced exon 7 inclusion in WT, mut-1, and mut-2 minigenes that harbor all or a subset of the SRSF1-binding motif sites, as compared to a control siRNA (siCt) (Figure 7D). The mut-3 minigene harbors neither GGA nor GGAGGA motif, and knockdown of SRSF1 had no effect on this minigene, as one would expect. Conversely, overexpression of SRSF1 increased inclusion of exon 7 in the mut-1 and mut-2 minigenes (Figure S7B). Moreover, we demonstrated that the regulatory effects of GGA and GGAGGA motifs through SRSF1 is specific, as knockdown or overexpression of another closely related SR protein family member SRSF2 had no effect on splicing of WT or mutant minigenes (Figure S7 A,B). Together, these results suggest that both GGA half sites and the canonical GGAGGA motif contributed to the overall inclusion activity of exon 7, with GGA half sites playing the dominant role.
To further validate our predictions, we similarly constructed HNRNPDL minigenes spanning exons 5 to 7 (a partial segment of intron 5 is deleted, see STAR Methods for detail; Figure 7F). The WT HNRNPDL exon 6 harbors eight GGA half sites and no GGAGGA motif. Through mutagenesis of the GGA motif sites in combination with knockdown and overexpression experiments, we confirmed that the GGA half sites strongly mediate exon 6 inclusion specifically through SRSF1, similar to what we observed for HNRNPD (Figure 7G,,HH and Figure S7C,D).
Discussion
The intrinsic flexibility of RBPs in recognizing their RNA regulatory sequences imposes a big challenge in accurate characterization and predictive modeling of their specificity, even when a large number of binding footprints are mapped by CLIP. To address this problem, we have developed a statistical model, named mCross, for de novo motif discovery using CLIP data. mCross builds on the critical observation that protein-RNA crosslinking in CLIP experiments frequently occurs at specific positions within the motif, which can be mapped at single-nucleotide resolution (Weyn-Vanhentenryck et al., 2014; Zhang and Darnell, 2011). These crosslink sites provide precise landmarks of motif sites in input sequences, and thus dramatically reduce the search space during de novo motif discovery.
We note that the proximity of RBP motif sites to potential crosslinking events was previously used as input in another algorithm, Zagros, to facilitate motif discovery (Bahrami-Samani et al., 2015). However, mCross and Zagros differ in several critical aspects. First, Zagros assumes UV crosslinking occurs near RBP binding sites, so it models the proximity of motif sites to diagnostic crosslinking events using a geometric distribution (a motif site with a closer crosslinking event receives higher weight than one with a more distal crosslinking event). This is probably, at least in part, because Zagros focuses more on analysis of PAR-CLIP data, which introduces photoactivatable nucleoside analogs (such as 4SU) for crosslinking (Hafner et al., 2010). However, this assumption is not accurate for other CLIP protocols that crosslink proteins and endogenous nucleosides, as we found that crosslinking can occur at very specific positions in the motif sites (e.g., G2 and G6 in the UGCAUG motif for Rbfox CLIP). Such position-specific crosslinking cannot be precisely characterized by the previous model. Therefore, mCross uses a position-specific weight crosslinking model, which allows it to take advantage of the single-nucleotide resolution for the first time. Second, Zagros uses potential diagnostic crosslinking events in individual CLIP tags, rather than robust crosslink sites. In the case of eCLIP (or iCLIP) data, it takes all CLIP tags and assumes the immediately upstream position as a crosslink site due to crosslinking-induced truncation during reverse transcription. In practice, only a fraction of CLIP tags are truncated, so this approach suffers from a low signal-to-noise ratio. To avoid this issue, mCross uses high-confidence crosslink sites with reproducible truncation in multiple CLIP tags identified by CITS analysis (Shah et al., 2017; Weyn-Vanhentenryck et al., 2014).
Therefore, mCross has the unique advantage by jointly modeling RBP sequence specificity and precise protein-RNA crosslink sites at specific motif positions at single-nucleotide resolution. This dramatically improves the accuracy of motif discovery (as demonstrated by our comparison with other methods that represent the current state of the art; see below). In addition, the positional specificity of UV crosslinking may also allow us to learn the underlying mechanisms of photocrosslinking between protein and RNA (e.g., what kind of contact facilitates UV crosslinking), although this is beyond the focus of this study.
We applied mCross to the largest CLIP datasets generated thus far by ENCODE to define motifs of 112 unique RBPs. Importantly, we also developed multiple quantitative measures to assess the reliability of the results. In particular, we performed genome-wide AI site analysis using CLIP data to detect SNPs affecting protein-RNA interactions, as these AI sites provide a large number of naturally occurring perturbation experiments in vivo that can be used to validate the accuracy of the discovered motifs. Our analyses suggest that mCross outperforms several other state-of-the-art methods we tested. In addition, when multiple, distinct motifs are discovered for the same RBP, from which ambiguity frequently arises, AI analysis also provides a means of selecting the most reliable motif. While we only used AI sites to validate the RBP motifs, it worth noting that AI sites filtered by PWMs also provided us with a subset of high-confidence SNPs that directly affect protein-RNA interactions, which may have functional implications in human populations. This list is particularly relevant given the observation that 90% of human disease- or trait-associated SNPs identified by genome-wide association studies (GWAS) are located in the noncoding regions of the genome (Hindorff et al., 2009), including introns, and are enriched in expression or splicing quantitative trait loci (eQTLs or sQTLs) (Li et al., 2016). SNPs modulating protein-RNA interactions may represent one important mechanism through which genetic variation can directly affect variation in gene expression and splicing.
We expect that the resulting list of motifs identified by mCross, complemented by an interactive, searchable web interface, will be a useful resource for the research community to make biological discoveries. In a recent study, we showed the importance of a distinct LIN28 (U)GAU motif discovered by mCross in differential binding by LIN28 and suppression of two subclasses of let-7 microRNAs that are major downstream targets of LIN28 (Ustianenko et al., 2018). In that case, crosslinking of LIN28 to the last uridine in the motif was also experimentally validated (Ransey et al., 2017). In this study, we found that SRSF1 binds clusters of GGA half sites, in addition to the previously characterized GGAGGA motif, as the predominant mode of target recognition. This flexibility implicates a much larger repertoire of target transcripts that were not previously appreciated. Indeed, we estimated that about 70–80% of SRSF1-regulated exons harbor only GGA clusters, but no canonical GGAGGA motif. Many of these previously unidentified targets may play important physiological roles in cells. To demonstrate this point, we focused on HNRNPD and HNRNPDL, and found that clusters of GGA half sites are critical for SRSF1 to activate inclusion of cassette exons in these genes. Intriguingly, it was previously demonstrated that these cassette exons encode GY-rich peptides, which can modulate the formation of multivalent high-order hnRNP assemblies and phase separation, and thereby the downstream splicing program on a global scale (Gueroussov et al., 2017). Combining previous results and our data presented in this study, we propose that SRSF1 may serve as an important upstream regulator of phase separation (Figure 7I). These case studies exemplify how improved definition of RBP binding specificity can lead to mechanistic insights into RNA regulation.
STAR Methods
EXPERIMENTAL MODEL AND SUBJECT DETAILS
CLIP data processing
We obtained CLIP data for Rbfox1–3 (Weyn-Vanhentenryck et al., 2014), Nova (Zhang et al., 2010), Ptbp2 (Licatalosi et al., 2012), Mbnl2 (Charizanis et al., 2012), and Lin28a (Cho et al., 2012) from separate studies. We previously analyzed Rbfox, Ptbp2 and Mbnl2 CLIP data part of the original studies using the CIMS package (Moore et al., 2014), a predecessor of CLIP Tool Kit (CTK) (Shah et al., 2017). Lin28a CLIP data was analyzed by the same pipeline (Moore et al., 2014). For each dataset, unique CLIP tags and mutations in unique tags, originally derived based on mapping to mm9, were liftOver to mm10. For CIMS analysis of deletions, we only included single-nucleotide deletions (and excluded deletions of two or more consecutive nucleotides).
eCLIP data of 70 RBPs in HepG2, 89 RBPs in K562, and 1 RBP in adrenal gland were downloaded from the ENCODE website (https://www.encodeproject.org; as of Dec 30, 2016) (Van Nostrand et al., 2017; Van Nostrand et al., 2016). In total, this dataset is composed of 112 unique RBPs, with 47 RBPs assayed in both HepG2 and K562 cells (Table S1). All mock control (input) data were also downloaded. The raw reads were processed to obtain unique CLIP tags mapped to hg19 using CTK (Shah et al., 2017), as described previously (Ustianenko et al., 2018). Only read2 (the read starting from 5ʹ end of the RNA tag) was used for analysis described in this paper. For each RBP, unique tags from the two replicates were combined for all analyses, except for evaluating reproducibility between the two replicates (see below). Significant CLIP tag clusters were called by requiring P<0.001 after Bonferroni multiple-test correction. Crosslinking-induced truncation sites (CITS) were called by requiring FDR<0.001. Crosslinking induced mutation sites (CIMS) were also examined but not reported in this paper because they appear to have low signal-to-noise ratio.
7-mer enrichment analysis
To provide seeds for de novo motif discovery using mCross, we performed 7-mer enrichment analysis using significant peaks with peak height (PH)≥10 tags. Peaks were extended for 50 nt on both sides relative to the center of the peak to extract the foreground sequences. Background sequences were extracted from the flanking regions of the same size (−550, −450) and (450, 550) relative to the peak center. Sequences with more than 20% of nucleotides overlapping with repeat masked regions were discarded. 7-mers were counted in repeat-masked foreground and background sequences, and the enrichment of each 7-mer in the foreground relative to the background was evaluated using a binomial test. A z-score (and a p-value) was derived for each 7-mer, and denoted raw z-score.
We noticed a general enrichment of certain 7-mers (such as G-rich elements) in many eCLIP experiments. To minimize potential experimental biases and non-specific protein-RNA interactions, we normalized the raw z-score of each 7-mer by subtracting median across all experiments followed by scaling using the median absolute deviation (MAD), a robust estimate of the standard deviation. The resulting score was denoted the normalized z-score and was used to rank and identify top 7-mers.
We developed an asymmetric enrichment score for each top 7-mer to evaluate their statistical significance, as an assessment whether an RBP has binding specificity. We argue that if there is no 7-mer that is significantly more enriched in the foreground than the background, the distribution of the normalized z-scores should be symmetric. On the other hand, if an RBP shows high affinity to (a relatively small subset of) specific 7-mers, it should show a heavy tail on the right side of the distribution. We therefore derived a false discovery rate (FDR) based on the symmetry of the null distribution.
for each z=x(>0). W(.) denotes the number of 7-mers satisfying the specified criterion.
The mCross model and the optimization algorithm
Most of the current de novo motif discovery tools (such as MEME (Bailey and Elkan, 1994) and HOMER (Heinz et al., 2010)) use a standard model of a position-specific weight matrix (PWM) to characterize the specificity of DNA- or RNA-binding proteins. Given a set of sequences bound by a specific RBP containing K binding sites of width W, the likelihood ratio of observing the data based on the motif model versus the background model can be written as follows:
where piB(k,i) and p0B(k,i) are the probability of observing base b= B(k,i) in position i of site k according to the motif and background models, respectively.
After log transformation and simple rearrangements,
The mCross model augments the standard PWM model by jointly modeling the RBP sequence specificity and the precise protein-RNA crosslink sites at specific motif positions at single-nucleotide resolution. Denote qi,k and q0 the probability of protein-RNA crosslinking at position i of the motif site k according to the motif and background models, respectively, the likelihood ratio of observing K sites of size W can be written as:
After rearrangement, the log likelihood ratio can be written as follows:
This model can be extended to allow crosslinking in specific positions outside the core motif:
where s = 1,2,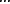 ,V indicates positions in the core motif or immediate flanking sequences (e.g., 2-nt extension on both sides of the core motif).
,V indicates positions in the core motif or immediate flanking sequences (e.g., 2-nt extension on both sides of the core motif).
To search for parameters pib and qi that optimize the objective function (we assume the background probabilities p0b = 0.25 and q0 = 1/V in this study), mCross currently uses a seed-based search strategy (Liu et al., 2002). In brief, a ranked list of top 7-mers is obtained based on their normalized z-score and asymmetric enrichment score (q<0.05; we limit to the top 10 if there are more than 10 7-mers with q<0.05). RBPs without asymmetrically enriched 7-mers are not analyzed.
To initiate motif search, mCross first groups top 7-mers with ≤2 mismatches without allowing shifts; each 7-mer group initiates one motif. Specifically, each 7-mer in a group is extended with the degenerate nucleotide ‘N’ for 2 nt on each side and then used as a seed to search for exact or inexact matches (≤m mismatches; m=1 for this study) around crosslink sites derived from CIMS or CITS analyses. These matches provide the list of all candidate RBP binding sites. Initially, all candidate sites are included to derive the motif model and calculate the log likelihood ratio L. An iterative procedure is then used to exclude or include each candidate site based on whether the adjustment improves the likelihood. The algorithm stops upon convergence or reaching the maximum iterations.
The objective function in eq. (6) in general favors degenerate motifs when more than one site is allowed in each input sequence. To reward more specific motifs, similar to (Liu et al., 2002, we introduced modifications to eq. (6) to generate results presented in this paper:
where
Among the 160 eCLIP experiments (with replicates combined), mCross discovered at least one motif for 144 experiments.
Motif clustering
For each RBP, we clustered similar motifs reported by mCross using Stamp (Mahony and Benos, 2007). PWMs were trimmed from both sides to remove positions with low information content ≤0.2, where information content was defined as in (Schneider et al., 1986). The Pearson correlation coefficient was adopted to measure the distance of each compared pair of PWMs. Local Smith-Waterman ungapped alignment and unweighted pair group method with arithmetic mean (UPGMA) were used to perform alignment and grow the cluster tree. The number of clusters was determined by minimizing the Calinski-Harabasz (CH) index provided by Stamp. If there was no global optimized cutoff using CH index, the clustering trees were cut at height 0.05.
Reproducibility of RBP sequence specificity
We developed rank-based measure of disconcordance of top 7-mer enrichment between the two replicates. To this end, we derived normalized z-score to rank 7-mers for each individual replicate. For the top T among a total of N=47=16,384 7-mers (T=20 for this study) with ranksum c= T(T + 1)/2 in replicate A, we obtained their rank sum in replicate B:
where rt(t = 1,…,T) is the rank of each top 7-mer.
Vice versa, for the top T 7-mer in replicate B, we obtained their rank sum ρA in replicate A. The disconcordance of the two replicates is measured by a score D.
We can similarly compare whether top 7-mers identified at CLIP tag cluster peaks are also ranked high in sequences near inferred crosslink sites. Denote the rank sum of the top 7-mers in sequences of crosslink sites ρ.
In this work, we consider an RBP with D<0.05 between the two replicates as having reproducible sequence specificity. We also used D-score to compare top 7-mers in peaks and CITS, and to compare CLIP and RNAcompete data (Ray et al., 2013).
SNP calling in HepG2 and K562 cells using eCLIP and whole genome sequencing data
If a protein-RNA interaction site is affected by genetic variation at a heterozygous SNP site, i.e., allelic interaction (AI), the two alleles will have different numbers of supporting CLIP tags. We used global analysis of AI sites to validate RBP motifs discovered by mCross.
To identify AI sites in CLIP data, we first called heterozygous SNPs in HepG2 and K562 cells using eCLIP (including mock) and whole genome sequencing (WGS) data. For each sample (either CLIP or mock) in the eCLIP dataset, the genomic mapping information of the unique tags was extracted, stored in a sam file, and converted to bam using SAMtools (Li et al., 2009). The bam files of all samples (including both CLIP and mock data) of the same cell line were merged together for HepG2 and K562, respectively. The variant-calling procedure was implemented following GATK (v3.8)’s best practice recommendations for RNA-seq data with minor modifications (McKenna et al., 2010). In particular, no “MarkDuplicate” step was carried out, as PCR duplicates have already been removed using a method optimized for CLIP data and the resulting unique tags were used as input. Per-base sequencing error was estimated by “BaseRecalibrator”. Then, we separately called variants for each cell line with “HaplotypeCaller” (with stand_call_conf set to 20 to ensure high sensitivity). Limited by the lack of truth/training sets required by the Variant Quality Score Recalibration (VQSR) step for eCLIP data, we adopted hard filters for variant filtration. Only bi-allelic SNPs with QualByDepth (QD) > 5 and depth (DP)>10 were kept. SNPs overlapping with RNA editing sites, as annotated in DARNED (Kiran and Baranov, 2010), were excluded.
To complement and improve genotype calls derived from eCLIP data, we performed variant calling using WGS data of HepG2 and K562 cells, respectively (Table S3), following the GATK “best practices” protocol for DNA-seq data. Briefly, for each cell line, WGS reads from different platforms were aligned to hg19 using BWA (Li and Durbin, 2010) and were merged together. We performed “MarkDuplicates” to remove PCR duplicates and used “BaseRecalibrator” to ensure the base quality. The variant calling step was carried out by “HaplotypeCaller” (with stand_call_conf set to 30). For variant filtering, we excluded variants on annotated RNA editing sites in DARNED (Kiran and Baranov, 2010) and applied the VQSR step to ensure that 99% of Hapmap SNPs in our data were included in the final call set. Only WGS SNPs covered by ≥10 eCLIP tags were used for genotype correction. We defined three subsets of SNPs to be considered in the following AI site analysis: 1) SNPs called heterozygous consistently in both eCLIP and WGS data. 2) the intersection of eCLIP and WGS data, but as heterozygous only in WGS data (the genotype call from WGS data was used for these SNPs); and 3) heterozygous SNPs called only in eCLIP data. We also filtered the last two categories by keeping only bi-allelic SNPs. SNPs called only from eCLIP data that were called as homozygous in WGS data or were inconstant with dbSNP (v138) genotypes were also excluded.
Finally, for each cell line and RBP, the number of unique CLIP tags supporting each allele of a heterozygous SNP was extracted from bam files with SAMtools (Li et al., 2009). We inferred the sense transcript strand of each SNP by pooling CLIP tags from all CLIP and mock data and counting the number of tags from each strand. The strand with a majority of supporting tags was considered the sense strand. Only heterozygous SNPs with unambiguously inferred transcript strand (i.e., #sense read/(#sense read+#antisense read)>0.9) were included in our analysis. The final dataset consisted of 229,265 and 155,388 heterozygous SNPs in HepG2 and K562 cells, respectively (Table S3).
Identification of allelic interaction sites from eCLIP data
To assess allelic binding of each RBP at a heterozygous SNP, we counted the number of sense CLIP tags of the RBP supporting each allele. Two types of control data were used for comparison: 1) pooled CLIP data of all other RBPs (except the RBP under consideration) in the same cell line (e.g., RBFOX2 vs. CLIP data of all other RBPs except RBFOX2); 2) all pooled mock data in the same cell line (e.g., RBFOX2 vs. mock). For each comparison, the magnitude of allelic imbalance was defined as |ΔA|=|AAFRBP-AAFcontrol|, where AAF (alternative allele frequency) was estimated from the number of sense CLIP tags supporting the alternative allele divided by the total number of sense CLIP tags overlapping with the SNP. The statistical significance of AI was evaluated using a Fisher’s exact test. For this analysis, we considered all heterozygous SNPs with coverage ≥10 sense tags. Sites with |ΔA|≥ 0.1 and p<0.05 in either of the two comparisons were called significant AI sites (Table S4).
Validation of PWMs using AI sites
For each PWM derived by mCross, we trimmed the flanking motif positions with information content≤0.4. For each AI site, the PWM score (Stormo, 2000) of the sequence associated with allele was calculated. Briefly, the reference and alternative allele sequences flanking each AI site of size 2W-1 were extracted, where W is the width of the trimmed PWM. We scanned the sequences and calculated the PWM scores of all possible motif sites:
where a indicates the reference or alternative allele, j=1, 2, …,W is the offset of the motif site and i is the position of the nucleotide in the motif site. For the background base composition, we used p0B(a,j,i)=0.25.
The PWM score was then normalized:
where M and m are the maximal and minimal possible scores of the PWM, respectively. The binding affinity of the RBP to allele a is estimated to be Ba = maxj Baj. The SNP is considered to affect RBP binding if max(BRef, BAlt) > α and |BRef − BAlt| > δ, where α and δ are thresholds to be determined. The AI site is denoted consistent AI site (with respect to the PWM) if the allele with higher binding affinity also has a larger number of supporting CLIP tags (e.g., ΔA>0 and BAlt > BRef); otherwise, the AI site is denoted inconsistent AI site. For each PWM of an RBP, we determined the number of all consistent AI sites NConst and the number of all inconsistent AI sites NInconst. If the PWM correctly characterized the binding specificity of the RBP, we would expect NConst > NInconst. The excess of consistent AI sites over inconsistent AI sites was performed using a one-sided Binomial test with a null hypothesis r = NConst/(NConst + NInconst) < 0.5. We denote a PWM AI-consistent PWM if p<0.05 and r>0.5. The depletion of consistent AI sites over inconsistent AI sites was also performed using a one-sided Binomial test with a null hypothesis r = NConst/(NConst + NInconst) > 0.5. We denote a PWM AI-inconsistent PWM if p<0.05 and r<0.5.
To determine the optimal thresholds of α and δ, we used a representative PWM (i.e., the first PWM) for each RBP and performed a grid search of parameters α and δ that maximized the number of AI-consistent PWMs. For analysis described in this paper, we used α = 0.8 and δ = 0.09 to determine AI-consistent PWMs, as the combination maximized the number of AI-consistent PWMs. With these determined thresholds, we then ranked all PWMs of each RBP based on the FDR using a single-sided binomial test (null hypothesis r<0.5; FDR derived from Benjamini correction for each RBP). The most significant PWM was selected as the best PWM for each RBP (Figure 5). We also used the best PWMs for each RBP to define the subset of individual consistent AI sites that most likely affect RBP binding directly (Table S4).
Comparison of AI-consistent PWMs identified by mCross and other de novo motif discovery programs
We compared the number of AI-consistent PWMs by mCross, MEME (Bailey and Elkan, 1994), DREME (Bailey, 2011), HOMER (Heinz et al., 2010), and Zagros (Bahrami-Samani et al., 2015), and RNAcompete (Ray et al., 2013). PWMs by MEME, DREME, HOMER and Zagros were derived from eCLIP data for all RBPs in HepG2 and K562 cells using 100 nt sequences around CLIP tag peaks as foreground. Flanking sequences of the same size (500 nt away from peaks) were used as background, if required. We adopted the following parameters for each program to limit the motif size to 4–7 nt, allowing any number of sites per sequence: MEME: -dna -mod zoops -nmotifs 10 -minw 4 -maxw 7; DREME: -dna -norc -m 10 -mink 4 -maxk 7; HOMER: -len 4,5,6,7 -S 10. For Zagros, we provided the coordinates of CLIP tags (with the immediately upstream position as potential diagnostic crosslinking events) together with the coordinate and sequences around CLIP tag peaks as input; the motif length is set as 6, which is the default. For comparison, only the first PWM reported by each program for each RBP was used. In addition, we also considered 18 RBPs assayed by eCLIP that have one or more PWMs from RNAcompete for comparison with mCross. The AI-consistent PWMs were defined as described above, using the same parameters.
Prediction of SRSF1 binding GGA clusters
We predicted clustered GGA motif sites that are bound by SRSF1 using mCarts (Weyn-Vanhentenryck and Zhang, 2016; Zhang et al., 2013). Briefly, we trained a HMM model using sequences centered at SRSF1 eCLIP tag cluster peaks, extending 50-nt flanking either side of every peak. Sequences without overlaps with CLIP tags were used as background. We trained one model for each cell type and predicted 1,781,913 and 1,759,031 clusters for HepG2 and K562, respectively. We found substantial overlap between the two models, with 1,685,575 overlapping clusters (Figure S6C) and highly correlated cluster scores (r=0.98; Figure S6D). As such, we decided to focus on the HepG2-generated model because of its larger training set. For comparison, we also predicted conserved GGAGGA sites using branch length score (BLS) estimated from multiple alignments of 40 mammalian species (Zhang et al., 2008).
Analysis of differential splicing upon SRSF1 knockdown
We downloaded RNA-seq data derived from SRSF1 shRNA knockdown and matched control cells for both cell lines from ENCODE (Van Nostrand et al., 2017). RNA-seq reads were mapped with OLego (v1.1.5) using the stranded mode (Wu et al., 2013). Alternative splicing quantification and differential splicing analysis upon SRSF1 knockdown were performed using the Quantas pipeline as previously described (Yan et al., 2015), requiring read coverage≥20 reads and FDR≤0.05. To generate RNA maps of SRSF1-dependent splicing, we used cassette exons with change in percent spliced in (PSI) |ΔΨ|≥0.2 in HepG2 cells (465 exons with SRSF1-dependent inclusion and 396 exons with SRSF1-dependent exclusion). For sensitivity and positive prediction value plots, we required |ΔΨ|≥0.1 to identify 373 exons with SRSF1-dependent inclusion in both HepG2 and K562 cells. Direct SRSF1 target cassette exons were predicted using overlapping GGA clusters and the exons were ranked by the cluster with the maximum motif score.
Construction of minigenes
We constructed HNRNPD wild-type (WT) minigene spanning exons 6 to 8 of human HNRNPD into pcDNA3.1(+) vector (Invitrogen) using BamHI and XhoI sites, respectively. This minigene includes partial sequences of intron 7 (150 nucleotides at the 5’ end and 150 nucleotides at the 3’ end, therefore a total of 300 nucleotides of intron 7). Artificial mutations were engineered into HNRNPD WT minigene using the QuikChange Site-Directed Mutagenesis Kit to generate HNRNPD mut-1, HNRNPD mut-2 and HNRNPD mut-3 minigenes, respectively. Similarly, we constructed HNRNPDL WT and mutant minigenes spanning exons 5 to 7 of human HNRNPDL using BamHI and XbaI sites, respectively. The HNRNPDL minigenes include partial sequences of intron 5 (150 nucleotides at the 5’ end and 150 nucleotides at the 3’ end, therefore total 300 nucleotides of intron 5). The absence of artifacts in all minigenes was confirmed by sequencing the entire inserts.
Cell culture and minigene transfection
HeLa cells were maintained in DMEM supplemented with 10% fetal bovine serum at 37°C in 5% CO2. Minigene plasmids (1.0 μg) were transfected in a well of a 6-well plate using X-tremeGENE DNA Transfection Reagents (Roche), according to manufacturer’s instructions. Total RNA was isolated 48 hr after the transfection.
RNA-interference and minigene transfection
siRNAs (50 nM) were transfected into HeLa cells using Lipofectamine™ RNAiMAX Reagent (Invitrogen) according to manufacturer’s instructions. After 24 hr of siRNA transfection, minigene plasmids were transfected using X-tremeGENE DNA Transfection Reagents (Roche) as described above. After 48 hr of minigene transfection, cells were harvested. Half of the cells were processed for total RNA isolation to perform RT-PCR and half of the cells were processed for total protein isolation to perform immunoblotting.
cDNA overexpression and minigene transfection
Construction of cDNA vectors, pCGT7-SRSF1 encoding T7-tagged human SRSF1 and pCGT7-SRSF2 encoding T7-tagged human SRSF2, was previously reported (Caceres et al., 1997). Transfection reaction in HeLa cells includes 0.3 μg of either pCGT7 empty vector (Vector) or pCGT7-SRSF1 or pCGT7-SRSF2, and 1 μg minigene in a well of a 6-well plate using X-tremeGENE DNA Transfection Reagents (Roche), according to the manufacturer’s instructions. Cells were harvested 48 hr after the transfection. Half of the cells were processed for total RNA isolation to perform RT-PCR and half of the cells were processed for total protein isolation to perform immunoblotting.
RNA isolation and RT-PCR
Total RNA was isolated using TRIzol® reagent, followed by DNaseI treatment (RQ1 RNase-Free DNase, Promega). Reverse transcription was performed using oligo-dT primer and ImProm-II™ reverse transcriptase (Promega). Radioactive PCR was conducted using 32P-a-dCTP, 1.25 units of AmpliTaq® (Invitrogen) and 26 cycles. Products were run on a 5% PAGE and the bands were quantified using a Typhoon FLA 7000 (GE Healthcare). Exon inclusion efficiency was calculated as PSI. To ensure minigene-specific splicing analysis, we used pcDNA3.1(+) vector-specific primers for amplification of both HNRNPD and HNRNPLD minigenes and their mutant derivatives.
Isolation of total protein and immunoblotting
To isolate total protein, cells were lysed using lysis buffer (0.2% NP-40, 200 mM NaCl, 50 mM Tris pH 7.4, 2 mM MgCl2, 1 mM DTT, 1 mM PMSF, 100 mM NaF, protease inhibitor cocktail) and passing through syringe. After gentle vortex, lysates were incubated on ice for 15 min. After centrifugation (15,000 × g, 10 min), the supernatants were collected as total cell extracts. Immunoblotting was performed using primary antibody and IRDye-680LT- or 800CW-labeled secondary antibodies for detection in a Li-Cor Odyssey.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-T7 | Cold Spring Harbor Laboratory | T7-KLH 42 1–87 |
| Mouse monoclonal anti-SRSF1 | Cold Spring Harbor Laboratory | AK-96 |
| Mouse monoclonal anti-SRSF2 | Gift from James Stévenin | NA |
| Rabbit polyclonal anti-β-Tubulin III | Genscript | A01203 |
| Experimental Models: Cell Lines | ||
| Human HeLa cells | ATCC | NA |
| Oligonucleotides | ||
| siRNA against Luciferase (used as control siRNA): 5′-GCCAUUCUAUCCUCUAGAGGAUGdTdT-3′ | (Cartegni et al., 2006) | NA |
| siSRSF1-N1 | SIGMA-ALDRICH | SASI_Hs01_00115056 |
| siSRSF1-N2 | SIGMA-ALDRICH | SASI_Hs01_00115062 |
| siSRSF2-N1: 5′-AAUCCAGGUCGCGAUCGAAdTdT-3′ | (Gabut et al., 2005) | NA |
| siSRSF2-N2: 5′-CACGAAGGUCCAAGUCCAAdTdT-3′ | (Cheng et al., 2016) | NA |
| Primers for amplification of HNRNPD and HNRNPDL minigenes: Forward: 5’- TAATACGACTCACTATAGGG-3’; Reverse: 5’-TAGAAGGCACAGTCGAGG-3’ | This study | NA |
| Software and Algorithms | ||
| CLIP data analysis by CTK | (Shah et al., 2017) | http://zhanglab.c2b2.columbia.edu/index.php/CTK |
| The MEME Suite | (Bailey and Elkan, 1994) | http://meme-suite.org/ |
| Stamp | (Mahony and Benos, 2007) | http://www.benoslab.pitt.edu/stamp/ |
| RNAcompete | (Ray et al., 2013). | https://github.com/morrislab/RNAcompete |
| Burrows-Wheeler Aligner | (Li and Durbin, 2010) | http://bio-bwa.sourceforge.net/ |
| Genome Analysis Toolkit | (McKenna et al., 2010) | https://software.broadinstitute.org/gatk/ |
| SAMtools | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| mCarts | (Weyn-Vanhentenryck and Zhang, 2016; Zhang et al., 2013) | https://zhanglab.c2b2.columbia.edu/index.php/MCarts_Documentation |
| OLego | (Wu et al., 2013) | https://zhanglab.c2b2.columbia.edu/index.php/OLego |
| Other | ||
| Rbfox1–3 CLIP data | (Weyn-Vanhentenryck et al., 2014) | SRP035321 |
| Nova CLIP data | (Zhang et al., 2010) | SRP002550 |
| Ptbp2 CLIP data | (Licatalosi et al., 2012) | SRP023497 |
| Mbnl2 CLIP data | (Charizanis et al., 2012) | SRP013558 |
| Lin28a CLIP data | (Cho et al., 2012) | GSE37114 |
| eCLIP data | (Van Nostrand et al., 2017; Van Nostrand et al., 2016) | https://www.encodeproject.org |
| SRSF1 shRNA-seq data | (Van Nostrand et al., 2017) | https://www.encodeproject.org |
| DARNED | (Kiran and Baranov, 2010) | http://darned.ucc.ie |
Supplementary Material
Supplemental Figures 1-7 and Legends
Supplemental Table 2
Table S2: Summary of mCross-discovered RBP motifs after clustering.
Related to Figure 3.
Supplemental Table 3
Table S3: SNP calling in HepG2 and K562 cells using whole-genome sequencing and eCLIP data.
Supplemental Table 4
Table S4: Summary of AI sites identified from eCLIP data.
Related to Figure 4.
Supplemental Table 5
Table S5: Summary of AI analysis for all PWMs discovered by mCross.
Related to Figure 5.
Acknowledgements
We thank James Stévenin for sharing SRSF2 antibody, Yingzhi Qian for assistance in RNA-seq data analysis, and members of the Zhang laboratory for helpful discussion. This study was supported by grants from the National Institutes of Health (NIH) (R01GM124486 and R03HG009528 to CZ). SB was in part supported by a Columbia Precision Medicine Research fellowship. High-performance computation was supported by NIH grants S10OD012351 and S10OD021764.
References
- Ascano M Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. [Europe PMC free article] [Abstract] [Google Scholar]
- Bahrami-Samani E, Penalva LO, Smith AD, and Uren PJ (2015). Leveraging cross-link modification events in CLIP-seq for motif discovery. Nucleic Acids Res 43, 95–103. [Europe PMC free article] [Abstract] [Google Scholar]
- Bailey T, and Elkan C (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol, 28–36. [Abstract] [Google Scholar]
- Bailey TL (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659. [Europe PMC free article] [Abstract] [Google Scholar]
- Caceres JF, Misteli T, Screaton GR, Spector DL, and Krainer AR (1997). Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J Cell Biol 138, 225–238. [Europe PMC free article] [Abstract] [Google Scholar]
- Cartegni L, Hastings ML, Calarco JA, Stanchina E.d., and Krainer AR (2006). Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet 78, 63–77. [Europe PMC free article] [Abstract] [Google Scholar]
- Charizanis K, Lee K-Y, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. (2012). Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75, 437–450. [Europe PMC free article] [Abstract] [Google Scholar]
- Chen M, and Manley JL (2009). Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 10, 741–754. [Europe PMC free article] [Abstract] [Google Scholar]
- Cheng Y, Luo C, Wu W, Xie Z, Fu X, and Feng Y (2016). Liver-specific deletion of SRSF2 caused acute liver failure and early death in mice. Mol Cell Biol 36, 1628–1638. [Europe PMC free article] [Abstract] [Google Scholar]
- Chi SW, Hannon GJ, and Darnell RB (2012). An alternative mode of microRNA target recognition. Nat Struct Mol Biol 19, 321–327. [Europe PMC free article] [Abstract] [Google Scholar]
- Chi SW, Zang JB, Mele A, and Darnell RB (2009). Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460, 479–486. [Europe PMC free article] [Abstract] [Google Scholar]
- Cho J, Chang H, Kwon SC, Kim B, Kim Y, Choe J, Ha M, Kim YK, and Kim VN (2012). LIN28A is a suppressor of ER-associated translation in embryonic stem cells. Cell 151, 765–777. [Abstract] [Google Scholar]
- Clery A, Sinha R, Anczukow O, Corrionero A, Moursy A, Daubner GM, Valcarcel J, Krainer AR, and Allain FHT (2013). Isolated pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proc Natl Acad Sci U S A 110, E2802–E2811. [Europe PMC free article] [Abstract] [Google Scholar]
- Cook KB, Kazan H, Zuberi K, Morris Q, and Hughes TR (2011). RBPDB: a database of RNA-binding specificities. Nucleic Acids Res 39, D301–D308. [Europe PMC free article] [Abstract] [Google Scholar]
- Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR, and Darnell RB (2005). Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev 19, 903–918. [Europe PMC free article] [Abstract] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, and Darnell RB (2001). Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499. [Abstract] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [Europe PMC free article] [Abstract] [Google Scholar]
- Darnell RB (2010). HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip Rev RNA 1, 266–286. [Europe PMC free article] [Abstract] [Google Scholar]
- Gabut M, Mine M, Marsac U, Brivet M, Tazi J, and Soret J (2005). The SR protein SC35 is responsible for aberrant splicing of the E1 alpha pyruvate dehydrogenase mRNA in a case of mental retardation with lactic acidosis. Mol Cell Biol 25, 3286–3294. [Europe PMC free article] [Abstract] [Google Scholar]
- Gueroussov S, Weatheritt RJ, O’Hanlon D, Lin ZY, Narula A, Gingras AC, and Blencowe BJ (2017). Regulatory expansion in mammals of multivalent hnRNP assemblies that globally control alternative splicing. Cell 170, 324–339 e323. [Abstract] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp A-C, Munschauer M, et al. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141. [Europe PMC free article] [Abstract] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [Europe PMC free article] [Abstract] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, and Manolio TA (2009). Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106, 9362–9367. [Europe PMC free article] [Abstract] [Google Scholar]
- Jankowsky E, and Harris ME (2015). Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol 16, 533–544. [Europe PMC free article] [Abstract] [Google Scholar]
- Jensen KB, Musunuru K, Lewis HA, Burley SK, and Darnell RB (2000). The tetranucleotide UCAY directs the specific recognition of RNA by the Nova K-homology 3 domain. Proc. Natl. Acad. Sci. USA 97, 5740–5745. [Europe PMC free article] [Abstract] [Google Scholar]
- Kiran A, and Baranov PV (2010). DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 26, 1772–1776. [Abstract] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, and Ule J (2010). iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17, 909–915. [Europe PMC free article] [Abstract] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [Europe PMC free article] [Abstract] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup GPDP (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [Europe PMC free article] [Abstract] [Google Scholar]
- Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, and Pritchard JK (2016). RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604. [Europe PMC free article] [Abstract] [Google Scholar]
- Licatalosi DD, and Darnell RB (2010). RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 11, 75–87. [Europe PMC free article] [Abstract] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469. [Europe PMC free article] [Abstract] [Google Scholar]
- Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, and Darnell RB (2012). Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 26, 1626–1642. [Europe PMC free article] [Abstract] [Google Scholar]
- Liu H-X, Zhang M, and Krainer AR (1998). Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 12, 1998–2012. [Europe PMC free article] [Abstract] [Google Scholar]
- Liu XS, Brutlag DL, and Liu JS (2002). An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol 20, 835–839. [Abstract] [Google Scholar]
- Long JC, and Caceres JF (2009). The SR protein family of splicing factors: master regulators of gene expression. The Biochemical journal 417, 15–27. [Abstract] [Google Scholar]
- Lunde BM, Moore C, and Varani G (2007). RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 8, 479–490. [Europe PMC free article] [Abstract] [Google Scholar]
- Mahony S, and Benos PV (2007). STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res 35, W253–258. [Europe PMC free article] [Abstract] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [Europe PMC free article] [Abstract] [Google Scholar]
- Moore M, Zhang C, Gantman EC, Mele A, Darnell JC, and Darnell RB (2014). Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat Protocols 9, 263–293. [Europe PMC free article] [Abstract] [Google Scholar]
- Ransey E, Björkbom A, Lelyveld VS, Biecek P, Pantano L, Szostak JW, and Sliz P (2017). Comparative analysis of LIN28-RNA binding sites identified at single nucleotide resolution. RNA Biol 14, 1756–1765. [Europe PMC free article] [Abstract] [Google Scholar]
- Ray D, Kazan H, Chan ET, Pena Castillo L, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, and Hughes TR (2009). Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol 27, 667–670. [Abstract] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. (2013). A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177. [Europe PMC free article] [Abstract] [Google Scholar]
- Sanford JR, Wang X, Mort M, VanDuyn N, Cooper DN, Mooney SD, Edenberg HJ, and Liu Y (2009). Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 19, 381–394. [Europe PMC free article] [Abstract] [Google Scholar]
- Schneider TD, Stormo GD, Gold L, and Ehrenfeucht A (1986). Information content of binding sites on nucleotide sequences. J Mol Biol 188, 415–431. [Abstract] [Google Scholar]
- Shah A, Qian Y, Weyn-Vanhentenryck SM, and Zhang C (2017). CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567. [Europe PMC free article] [Abstract] [Google Scholar]
- Singh R, and Valcarcel J (2005). Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol 12, 645–653. [Abstract] [Google Scholar]
- Stormo GD (2000). DNA binding sites: representation and discovery. Bioinformatics 16, 16–23. [Abstract] [Google Scholar]
- Tacke R, and Manley JL (1995). The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 14, 3540–3551. [Europe PMC free article] [Abstract] [Google Scholar]
- Ule J, Jensen K, Mele A, and Darnell RB (2005). CLIP: A method for identifying protein-RNA interaction sites in living cells. Methods 37, 376–386. [Abstract] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, and Darnell RB (2003). CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215. [Abstract] [Google Scholar]
- Ustianenko D, Chiu H-S, Treiber T, Treiber N, Weyn-Vanhentenryck SM, Meister G, Sumazin P, and Zhang C (2018). LIN28 selectively modulates a subclass of let-7 microRNAs. Mol Cell 71, 271–283.e275. [Europe PMC free article] [Abstract] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Blue SM, Dominguez D, Cody NAL, Olson S, Sundararaman B, et al. (2017). A large-scale binding and functional map of human RNA binding proteins. bioRxiv 10.1101/179648. [Europe PMC free article] [Abstract] [CrossRef] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Meth 13, 508–514. [Europe PMC free article] [Abstract] [Google Scholar]
- Wang Z, Kayikci M, Briese M, Zarnack K, Luscombe NM, Rot G, Zupan B, Curk T, and Ule J (2010). iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol 8, e1000530. [Europe PMC free article] [Abstract] [Google Scholar]
- Weyn-Vanhentenryck S, Mele A, Sun S, Yan Q, Farny N, Zhang Z, Xue C, Silver PA, Zhang MQ, Krainer AR, et al. (2014). HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 6, 1139–1152. [Europe PMC free article] [Abstract] [Google Scholar]
- Weyn-Vanhentenryck SM, and Zhang C (2016). mCarts: genome-wide prediction of clustered sequence motifs as binding sites for RNA-binding proteins. Methods Mol Biol. 1421, 215–226. [Abstract] [Google Scholar]
- Wilbert Melissa L., Huelga Stephanie C., Kapeli K, Stark Thomas J., Liang Tiffany Y., Chen Stella X., Yan Bernice Y., Nathanson Jason L., Hutt Kasey R., Lovci Michael T., et al. (2012). LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol Cell 48, 195–206. [Europe PMC free article] [Abstract] [Google Scholar]
- Wu J, Anczukow O, Krainer AR, Zhang MQ, and Zhang C (2013). OLego: Fast and sensitive mapping of spliced mRNA-Seq reads using small seeds. Nucleic Acids Res 41, 5149–5163. [Europe PMC free article] [Abstract] [Google Scholar]
- Yan Q, Weyn-Vanhentenryck SM, Wu J, Sloan SA, Zhang Y, Chen K, Wu JQ, Barres BA, and Zhang C (2015). Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc Natl Acad Sci U S A 112, 3445–3350. [Europe PMC free article] [Abstract] [Google Scholar]
- Zhang C, and Darnell RB (2011). Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat Biotechnol 29, 607–614. [Europe PMC free article] [Abstract] [Google Scholar]
- Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, and Darnell RB (2010). Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329, 439–443. [Europe PMC free article] [Abstract] [Google Scholar]
- Zhang C, Lee K-Y, Swanson MS, and Darnell RB (2013). Prediction of clustered RNA-binding protein motif sites in the mammalian genome. Nucleic Acids Res 41, 6793–6807. [Europe PMC free article] [Abstract] [Google Scholar]
- Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, and Zhang MQ (2008). Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev 22, 2550–2563. [Europe PMC free article] [Abstract] [Google Scholar]
Full text links
Read article at publisher's site: https://doi.org/10.1016/j.molcel.2019.02.002
Read article for free, from open access legal sources, via Unpaywall:
http://www.cell.com/article/S1097276519300929/pdf

Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Article citations
DDX RNA helicases: key players in cellular homeostasis and innate antiviral immunity.
J Virol, 98(10):e0004024, 30 Aug 2024
Cited by: 0 articles | PMID: 39212449 | PMCID: PMC11494928
Review
 Free full text in Europe PMC
Free full text in Europe PMCMass Spectrometric Evidence for the Under-represented RNA-Protein Cross-Links.
ACS Omega, 9(40):41863-41866, 25 Sep 2024
Cited by: 0 articles | PMID: 39403302 | PMCID: PMC11472617
Protocol to study the direct binding of proteins to RNA:DNA hybrids or RNA-DNA chimeras in living cells using cross-linking immunoprecipitation.
STAR Protoc, 5(3):103292, 12 Sep 2024
Cited by: 0 articles | PMID: 39264804 | PMCID: PMC11422765
splicekit: an integrative toolkit for splicing analysis from short-read RNA-seq.
Bioinform Adv, 4(1):vbae121, 17 Aug 2024
Cited by: 0 articles | PMID: 39219843 | PMCID: PMC11364168
Deciphering 3'UTR Mediated Gene Regulation Using Interpretable Deep Representation Learning.
Adv Sci (Weinh), 11(39):e2407013, 19 Aug 2024
Cited by: 1 article | PMID: 39159140 | PMCID: PMC11497048
Go to all (59) article citations
Data
Data behind the article
This data has been text mined from the article, or deposited into data resources.
BioStudies: supplemental material and supporting data
GEO - Gene Expression Omnibus
- (1 citation) GEO - GSE37114
Nucleotide Sequences (3)
- (1 citation) ENA - SRP013558
- (1 citation) ENA - SRP023497
- (1 citation) ENA - SRP002550
Protein structures in PDBe
-
(1 citation)
PDBe - 2m8dView structure
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
hnRNP A1 and the SR proteins ASF/SF2 and SC35 have antagonistic functions in splicing of beta-tropomyosin exon 6B.
J Biol Chem, 279(37):38249-38259, 18 Jun 2004
Cited by: 59 articles | PMID: 15208309
Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP).
Nat Methods, 13(6):508-514, 28 Mar 2016
Cited by: 716 articles | PMID: 27018577 | PMCID: PMC4887338
Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins.
Genome Biol, 21(1):90, 06 Apr 2020
Cited by: 112 articles | PMID: 32252787 | PMCID: PMC7137325
Characterization of RNA-Protein Interactions: Lessons from Two RNA-Binding Proteins, SRSF1 and SRSF2.
Methods Mol Biol, 1421:1-13, 01 Jan 2016
Cited by: 1 article | PMID: 26965252
Review
Funding
Funders who supported this work.
NHGRI NIH HHS (1)
Grant ID: R03 HG009528
NIGMS NIH HHS (2)
Grant ID: R01 GM042699
Grant ID: R01 GM124486
NIH (2)
Grant ID: S10OD012351
Grant ID: S10OD021764
NIH HHS (2)
Grant ID: S10 OD021764
Grant ID: S10 OD012351


