Abstract
Free full text
Biomarkers for epithelial-mesenchymal transitions
Abstract
Somatic cells that change from one mature phenotype to another exhibit the property of plasticity. It is increasingly clear that epithelial and endothelial cells enjoy some of this plasticity, which is easily demonstrated by studying the process of epithelial-mesenchymal transition (EMT). Published reports from the literature typically rely on ad hoc criteria for determining EMT events; consequently, there is some uncertainty as to whether the same process occurs under different experimental conditions. As we discuss in this Personal Perspective, we believe that context and various changes in plasticity biomarkers can help identify at least three types of EMT and that using a collection of criteria for EMT increases the likelihood that everyone is studying the same phenomenon — namely, the transition of epithelial and endothelial cells to a motile phenotype.
In the early 19th century, building on observations of microscopists now ancient, Schleiden and Schwann formulated the doctrine that cells are building blocks for plant and animal tissues (1). By the mid-19th century, Raspail, Remak, and Virchow expanded this hypothesis by suggesting that all cells come from preexisting cells — the so-called cell theory. Although referring to cell division, this now classic notion is prescient of another contemporary twist in the biology of cell maturation: beyond lineage development and normal differentiation, mature epithelial cells under new environmental pressures exhibit a local plasticity that allows them to morph into other mature phenotypes with or without proliferation (2, 3). Growing interest in the biology of these cellular transitions helped both establish epithelial cell plasticity as a field of study in the late 20th century and fashion much of the current thinking regarding morphogenesis in early embryonic development, tissue repair, and cancer metastasis (4–6). The details of some of these processes are not discussed here, as they are outlined in other articles in this Review Series on epithelial-mesenchymal transition (EMT) (7, 8). Instead, we offer a personal view gathered from our own experience and the literature regarding an approach documenting EMT events in culture or tissue. We hope this serves to stimulate other points of view as new data emerge.
Epithelial cell plasticity
The notion of epithelial cell plasticity derives largely from early studies of tissue regeneration in primitive experimental models. These models involve a variety of tissues, many of which have an epithelial component. For example, both amputated and injured structures from Urodela (newts and salamanders) regenerate damaged parts rather than scarring over wounds. Regenerative plasticity in these species is seen in limbs, facial bones, and tails; along the spinal cord and in the eye (retina, lens, and optic nerve); and in organs such as the intestinal tract and heart (9, 10). Such plasticity has been rigorously studied in the primitive epithelial lens (10, 11), probably because the eye of newts is a site of immunologic privilege — where immune responses are constrained and tissues protected from inflammation and scarification (9, 10). The lizard represents a phylogenic transition toward more complex organisms, as its tail (12, 13), but not limbs (14), first scar and then regenerate blastema to replace the missing appendage. Evidence for tissue regeneration in primates is scarce, except in the liver, occasional peripheral neurons, and the finger tips in limited circumstances (15). Wounds that are severe or caused by persistent injury typically heal as fibrotic scars.
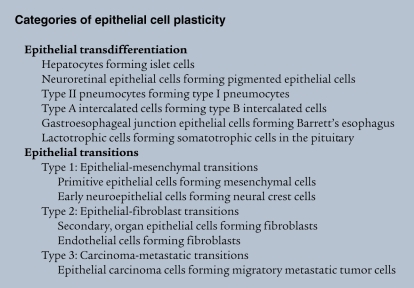
In mammals, experimental work on epithelial cell plasticity mainly follows the trail of two broad interests, metaplasia and EMT (Figure (Figure11 and Categories of epithelial cell plasticity). With regard to EMT, it is important to note that it was agreed in 2003, at the first meeting of The EMT International Association (TEMTIA), in Port Douglas, Australia, that epithelial-mesenchymal transformation and epithelial-mesenchymal transdifferentiation would be called epithelial-mesenchymal transition going forward.

Transdifferentiation generally refers to a process whereby one mature epithelial cell phenotype converts into a different mature epithelial cell, with or without cell division. It is unclear whether transdifferentiation involves a transition state. Three types of EMT are recognized depending on the phenotype of the output cells. Type 1 EMT is seen when primitive epithelial cells transition into mesenchymal cells that form the diaspora of the basic body plan following gastrulation or neural crest migration. These mesenchymal cells either undergo MET to form secondary epithelial cells or apoptose. Type 2 EMT is seen when secondary epithelial cells or endothelial cells populate interstitial spaces with resident or inflammation-induced fibroblasts, the latter during persistent injury. Type 3 EMT is part of the metastatic process, whereby epithelial tumor cells leave a primary tumor nodule, migrate to a new tissue site, and reform as a secondary tumor nodule.
Metaplasia is the conversion of one differentiated cell type into another, as observed in experimental tissue remodeling and random pathological tissue specimens, where it occurs spontaneously. Metaplasia is something cell biologists now call transdifferentiation. Evidence of transdifferentiation in mammalian cells is not as common as in amphibians but has been observed among islet cells, hepatocytes, lactotrophs, pneumocytes, and intercalated tubular cells (5, 16).
EMT involves the formation of motile cells from parent epithelial cells that are not themselves motile. Discrete, age- or stage-dependent conversion of primitive epithelial cells into motile cells by EMT is seen in early embryogenesis, when it is involved in gastrulation and neural crest migration. Later in organogenesis, the expression of local developmental cytokines drives secondary epithelial cells and endothelial cells (specialized squamous epithelial cells) to become fibroblasts through EMT. The fibroblasts generated by this process form endocardial cushions; close the cranial plates, facial bones, and mid-line palate; and populate interstitial spaces with resident fibroblasts as part of the normal expansion of connective tissues. In adult life, the cytokine bath generated by persistent tissue injury drives the formation of an abundance of fibroblasts by the same mechanism (4, 5, 17). EMT is also seen in subsets of carcinoma cells undergoing phenotypic conversion for invasion and metastasis (18, 19), suggesting that epithelial carcinomas create motile cancer cells by reusing some part of the molecular EMT program that normally serves the formation of adult fibroblasts (20).
Different types of EMT
The field of EMT today is vastly more expansive in scope and understanding than it was just a few years ago, particularly with new work on the role of EMT in tissue fibrosis and cancer metastasis (4, 6, 21). The study of various model systems involving an abundance of different epithelial cell types, often examined in culture and out of biological context, lends considerable uncertainty to the nature of common signaling and transcriptional pathways predictive of EMT. This is particularly true when one tries to compare mRNA pools generated under various experimental conditions. In March 2008, at an EMT meeting at Cold Spring Harbor Laboratory, we and others (8) suggested there is heuristic value in parsing EMT into three general subtypes based simply on the context under which they occur (Figure (Figure11 and Categories of epithelial cell plasticity). Type 1 EMT involves primitive epithelial cells transitioning to motile mesenchymal cells as part of gastrulation and primitive neuroepithelial cells generating migrating neural crest cells. In both situations, some of the cells generated by EMT are re-induced as secondary epithelial cells in mesodermal and endodermal organs by mesenchymal-epithelial transition (MET). Type 2 EMT involves secondary epithelial or endothelial cells transitioning to resident tissue fibroblasts. In mature tissues, these fibroblasts are induced in response to persistent inflammation. Type 3 EMT involves epithelial carcinoma cells in primary nodules transitioning to metastatic tumor cells in order to migrate through the blood stream and, in some cases, form secondary nodules in distant metastatic sites by MET.
When the process of EMT is looked at in this way, it is important to preserve the possibility of common mechanisms, recognizing that the three subtypes of EMT have some phenotypic similarities, but also many differences. For example, it is likely that mRNA pools from each subtype of input cells (primitive, secondary, and tumor epithelial cells) are considerably different, with many unshared transcripts. Likewise, the output cells (mesenchymal cells, fibroblasts, and metastatic tumor cells, respectively) have little in common other than the ability to move and are likely to express different pools of mRNA. We believe that parsing EMT into three subtypes will encourage comparisons among similar models and help identify better candidate small molecules predictive or instructive of type-specific transitions.
EMT resulting in either mesenchymal cells or fibroblasts
A principal distinction between the EMT involving primitive epithelial cells and that involving secondary epithelial cells is that type 1 EMT during embryogenesis produces mesenchymal cells, whereas type 2 EMT in adult or maturing tissues results in fibroblasts. From a historical perspective, little serious thought has been given to the origin of fibroblasts and their relation to mesenchymal cells. Primitive epithelial cells in Metazoans form primary mesenchymal cells by type 1 EMT during gastrulation (22, 23). These mesenchymal cells disperse and are variably induced, through MET, to form secondary epithelial cells that support early organogenesis. Unused mesenchymal cells that remain undergo programmed cell death (24). Coincident with organogenesis and the appearance of secondary (MET-derived) epithelial cells, fibroblasts first appear in mice after E9 following type 2 EMT in maturing tissues (25, 26). Because fibroblasts move, have front end–back end polarity, elongate with filopodia, and look like mesenchymal cells (17), and because we tend to label the two cell types interchangeably — particularly when referring to transitions in adult tissues — both cells are understandably and unfortunately mislabeled in the biomedical literature.
Other than the fact that mesenchymal cells have a shape that resembles fibroblasts and, like fibroblasts, express fibronectin and fibrillar collagens, there is no evidence that fibroblasts are of direct lineage descent from primitive mesenchymal cells. Tissue fibrosis, for example, does not occur during early embryogenesis, at least not until organogenesis begins (25) and the immune system reaches a functional level of maturity (27). Consistent with this observation is the fact that most mammalian tissues enjoy less immunologic privilege than the tissues of species such as Urodela and the realization that primitive mesenchymal cells are not likely to be true fibroblasts (25); that is, early embryos do not scar (28), and, unlike fibroblasts, primary mesenchymal cells are multipotent (29, 30), susceptible to MET (31), and do not express proteins that uniquely define fibroblasts (25).
It is only once fibroblasts formed by type 2 EMT materialize in the interstitial spaces between developing secondary epithelium and endothelium that these tissues are primed for an inducible fibrogenic response consequent to persistent inflammation. Fibroblast formation and fibrosis, therefore, are principally a feature of immunologic and tissue maturation. More recently, it has been observed that fibroblasts harvested from various tissues have distinct gene profiles (32) and behave differently following exposure to assorted matrices and cytokines (33). This is entirely consistent with the topographic distribution of variable pools of mRNA in fibroblasts, perhaps based on a sequential Hox gene code instructing differences in mRNA pools according to regions of the body plan (32, 34). Although this later notion has been interpreted through the narrow prism of mesenchymal cell ancestry, it is also completely consistent with the notion that fibroblasts formed by EMT differentially express genes or signaling pathways dependent on their previous life as mature epithelial or endothelial cells, and theoretically could be as heterogeneous as their parent cells.
EMT involving metastatic cancer cells
The concept that EMT involves the formation of metastatic cancer cells is based on the observation that acquisition of mesenchymal markers such as vimentin or S100A4 (also known as fibroblast-specific protein 1 [FSP1]) by epithelial carcinoma cells is associated with increased metastatic potential (18), as is nuclear overexpression of β-catenin (35) and loss of epithelial cell adhesion molecules such as E-cadherin (18, 20, 36–38). Type 3 EMT, while sharing some phenotypic similarities with other types of EMT, also may invoke the expression of more primitive markers suggestive of stem cells (39). Two recent interesting points about EMT producing fibroblasts can also be made in the setting of cancer. First, fibroblasts adjacent to primary epithelial tumor nodules share some genetic mutations with the tumor cells, suggesting that type 2 EMT forming fibroblasts occurs prior to the full onset of tumorigenesis (40, 41). On the other hand, studies comparing mutations in cancer cells and cancer-associated fibroblasts isolated from distant metastatic tumor tissue fail to demonstrate a cancer cell origin for these fibroblasts (42), suggesting that once epithelial tumor cells emerge, they no longer are a source of fibroblasts. Rather, the cancer-associated fibroblasts in metastatic sites are likely to be either attracted to a secondary tumor nodule from the local resident population or derived from endothelial sprouts that form neovascular vessels as the secondary nodules enlarge (43). Second, whereas type 2 EMT involves the transition of adult epithelial or endothelial cells into fibroblasts, type 3 EMT implies something different. Type 3 EMT is not a mechanism for forming fibroblasts, but a process that uses this mechanism to transition and ready nodular tumor epithelial cells for movement, invasion, and metastasis (20).
We review here some thoughts on criteria and common biomarkers for EMT in all three subtypes. Truly remarkable advances have been made in unraveling this interesting phenomenon of phenotypic transition, particularly in the generation of fibroblasts.
In vitro and in vivo criteria for type 2 EMT producing fibroblasts
Since the in vitro demonstration of EMT by urodele corneal, notochord, limb, and lens epithelial and endothelial cells in collagen gel immersion experiments in 1982 (44), much interest has focused on how to better detect epithelial cells undergoing EMT to become fibroblasts in different culture systems and tissues. Although detection of a change in phenotype or the measurement of movement is possible during EMT in culture, these are more difficult to recognize in tissues. Indeed, one of the historical problems in studying fibroblast lineage during EMT has been the lack of highly specific markers. In 1995, we reported from genetic screens that all fibroblasts express FSP1 and are likely to appear through type 2 EMT (26). This transition was demonstrated more convincingly in lineage-tracing studies during the formation of fibroblasts in renal tissues (45). Subsequent lineage-tracing studies confirmed these findings in other organs, including liver, lung, and heart (46–48). Of special interest, recently we found that endothelial cells also undergo type 2 EMT to form FSP1+ fibroblasts in heart (47) and kidney (49). Both epithelial and endothelial cells are likely to be substrates for new fibroblasts in adult tissue. A commonly used marker of active fibroblasts is α-SMA (50), but it is not specific for fibroblasts (51), as only a subset of fibroblasts express this protein (4). Elizabeth Hay (17) also points out that mesenchymal cells and fibroblasts should not be defined on the basis of α-SMA stress fibers, as such myofibroblast phenotypes are not thought to actively migrate. Insights from FSP1 studies used to detect type 2 EMT in vitro and in vivo have also served as a basis for studies of type 3 EMT involving cancer cells (20). New lineage-tracing technology in transgenic mice may allow this to be mapped in vivo (52).
Published studies often use somewhat random or ad hoc criteria for identifying EMT. The field has continued to grow, with nearly 600 articles now published on this subject. As we discuss here, from this work derive a few useful parameters for recognizing EMT either in cell culture or in tissues (see In vitro criteria for EMT and In vivo criteria for EMT). We believe that using a collection of criteria for EMT increases the likelihood that everyone is studying the same phenomena experimentally regardless of EMT type.
In vitro EMT.
In vitro evidence for type 2 EMT is suggested by the transition of defined epithelial cells into fibroblasts over a few days of culture. It has been known for decades that cultures of primary mammalian epithelial cells are at risk for contamination by fibroblast overgrowth, even if the culture was derived from a single epithelial cell. This occurrence of spontaneous type 2 EMT in culture is variable, and we suggest it is probably a function of unpredictable amounts of cytokines in different batches of fetal calf serum, the likely culprit being TGF-β. In controlled type 2 EMT experiments, using cultures of tubular epithelial cells, the addition of TGF-β (3 ng/ml) plus EGF (10 ng/ml) to serum-free medium produces consistent rates of type 2 EMT that are completed by 5 days (53, 54). Early removal of the ligands that induce type 2 EMT in some culture systems allows restoration of the epithelial cell phenotype, something known as reversible scatter (4), suggesting that the plasticity between type 2 EMT and MET is dynamic and can be pushed in either direction (5). Classically, active push back toward MET in mice occurs in the presence of bone morphogenetic protein 7 (BMP-7) (55).

Epithelial cells transitioning to fibroblasts lose polarity; increase their nuclear expression of several transcription factors (see below); lose the expression of E-cadherin, syndecan-1, and zona occludens 1 (ZO-1); increase their synthesis of cytoskeletal proteins (see below); rearrange actin stress fibers; become spindle-shaped; and start to move. Only later and variably do fibroblasts express α-SMA as myofibroblasts (see In vitro criteria for EMT). Transitioning epithelial cells are also resistant to apoptosis (56, 57). The in vitro migration of new fibroblasts following type 2 EMT is the expected mature function of this phenotypic transition (53, 58–60). The fibroblasts formed should also show a stable phenotype on removal of the induction stimulus and with passage or time.

In vivo EMT.
In vivo evidence for type 2 EMT is more difficult to document, because many of the tissue landmarks are disrupted by inflammation and cicatrization. In experimental systems, the cleanest approach to identify type 2 EMT is to use reporter genes in epithelial and endothelial cells that can be tracked in new fibroblasts on transition (45–48). When marking studies are not possible, such as in human tissues, epithelial or endothelial cell expression of FSP1 adjacent to a disrupted basement membrane, occasionally associated with cell elongation toward the interstitial spaces, is suggestive (26). Some FSP1+ epithelial cells in this transitional environment should show new expression of HSP47 (51) or collagen type 1, the latter being much more difficult to demonstrate, and partial to complete loss of E-cadherin and cytokeratin (4). α-SMA can be seen in mature fibroblasts but is not expected in newly transitioning epithelial cells (17). Transitioning epithelial cells normally exhibit new expression of transcription factors (61–63) and nuclear localization of CArG box–binding factor–A (CBF-A) (54) and complexes of β-catenin and lymphoid enhancer–binding factor (LEF) (64). EMT by definition is a plastic process with various intermediate stages, and it is likely that the changing markers mentioned above are present to a varying degree in all three subtypes of EMT (see In vivo criteria for EMT).
Biomarkers of EMT
A variety of biomarkers have been used to demonstrate all three subtypes of EMT. Here we examine a few of the more common markers, some of which are acquired and some of which are attenuated during transition (Table (Table1). 1).
Table 1
Markers of EMT

Cell-surface markers of EMT.
A change in expression of E-cadherin is the prototypical epithelial cell marker of EMT. E-cadherin is expressed in epithelial cells, and its expression is decreased during EMT in embryonic development, tissue fibrosis, and cancer (65). Moreover, loss of E-cadherin function promotes EMT (4, 66). In recent years, changes in the level of expression of different cadherins, so-called cadherin switches, have been increasingly used to monitor EMT. Indeed, the cadherin switch from E-cadherin to N-cadherin, which is expressed in mesenchymal cells, fibroblasts, cancer cells, and neural tissue, has often been used to monitor the progress of EMT during embryonic development and cancer progression. In addition, because OB-cadherin is a more definitive marker for activated fibroblasts, an E-cadherin–OB-cadherin switch is of interest for type 2 EMT associated with fibrogenesis (67).
EMT is associated classically with a relocation of cells from a basement membrane microenvironment into a fibrillar ECM. A change in the level of expression of different integrins (an integrin switch) often reflects alterations in cell-ECM interactions. In addition, integrin signaling facilitates EMT (68, 69), and various integrins are expressed on both epithelial and mesenchymal cells. As a result, integrins in general have limited utility as generalized biomarkers for EMT. There are, however, examples where integrins can be used as EMT markers in a context-dependent manner. In colon carcinoma, only cancer cells that have undergone type 3 EMT to a metastatic phenotype express high levels of β6 integrin (normal epithelial cells and noninvasive cancer cells have low-level expression) (70). During gastrulation, type 1 EMT is associated with de novo expression of α5β1, which is a receptor for fibronectin (71). Similarly, type 2 EMT in experimental kidney fibrosis is associated with increased α5 integrin expression (72). Increased expression of α5 integrin also correlates with the metastatic potential of B16F10 melanoma cells and EMT (73), suggesting that α5 integrin plays a role in each subtype of EMT.
Another EMT marker that reflects adaptation to the altered ECM microenvironment associated with EMT is the collagen-specific receptor tyrosine kinase DDR2 (discoidin domain receptor tyrosine kinase 2) (74). Upon binding to type I or type X collagen, DDR2 mediates upregulation of MMP1 and cell motility (74, 75). In adult tissues, expression of DDR2 is confined to subsets of fibroblasts and vascular smooth muscle cells, whereas de novo expression of DDR2 in endothelial cells is associated with type 2 EMT (47, 76). DDR2 expression in cancer cells correlates with increased invasiveness, demonstrating its utility in identifying type 3 EMT (74, 77).
Cytoskeletal markers of EMT.
FSP1 is a member of the family of Ca2+-binding S100 proteins (26). It is a prototypical fibroblast marker for detecting EMT in cancer and fibrogenesis (20, 45). In tissue fibrosis, most epithelial cells undergoing type 2 EMT express FSP1 early in transition to fibroblasts, and lineage tagging in transgenic reporter mice reveals that more than one-third of all FSP1+ fibroblasts in fibrotic kidneys and livers are EMT derived (45, 48). In addition, in models of cancer, metastatic cells often express FSP1 as part of the molecular program of type 3 EMT (20). Ectopic expression of FSP1 itself facilitates EMT in adult epithelial cells and cancer cells (20, 45, 53). In embryonic development, FSP1 is expressed from E8.5 in the mouse, well after mesenchymal cells first appear (26). Because FSP1 is mainly confined to transitioning epithelial cells and fibroblasts as opposed to primitive mesenchymal cells, it has limited utility for detection of type 1 EMT.
A controversial marker of EMT is the intermediate filament vimentin, which is expressed in various cells, including fibroblasts, endothelial cells, cells of the hematopoietic lineages, and glial cells (78, 79). During embryonic development in the mouse, vimentin is first expressed at E6.5–E7.0, and it is used as a marker for type 1 EMT during gastrulation (80, 81). However, because adult epithelial cells transiently express vimentin in response to various insults (82), vimentin should not be considered a marker of type 2 EMT in the setting of fibrosis. By contrast, vimentin is commonly used to identify cells undergoing type 3 EMT in cancers (63, 83). This is based on a positive correlation of vimentin expression with increased invasiveness and metastasis (84).
α-SMA is one of six actin family members. In the adult, prominent α-SMA expression can be found in vascular smooth muscle cells and myoepithelial cells (85). During embryonic development, the EMT that gives rise to the endocardial cushion is characterized eventually by de novo α-SMA expression (86). Type 2 EMT, which contributes to tissue fibrosis, is also sometimes associated with cells that eventually express α-SMA as myofibroblasts. Although some of these cells that express both α-SMA and FSP1 are known as myofibroblasts, most α-SMA+ cells express only α-SMA and are quite distinct from EMT-derived fibroblasts (43, 47, 51). In cancer, evidence that type 3 EMT is associated with α-SMA is mostly confined to breast cancer (87), where α-SMA largely is detected in breast tumors of the “basal phenotype” (88).
β-Catenin is a cytoplasmic plaque protein that plays a dual role in EMT: it links cadherins to the cytoskeleton and serves as a cotranscriptional activator together with T cell factor (TCF)/LEF (89). β-Catenin activity is mainly regulated by mechanisms controlling the level of β-catenin in the cytoplasm, through either its recruitment to cadherin-binding partners or ubiquitination and subsequent degradation (90). The β-catenin/TCF/LEF complex directly controls gene expression associated with EMT, particularly Snail1 (91). β-Catenin has been used as a marker of EMT in various studies of embryonic development, cancer, and fibrosis (4, 35, 92). Although β-catenin is localized to cell membranes in normal epithelial cells and noninvasive tumor cells, it is located either in the cytoplasm (something that is reflective of dissociation from E-cadherin) or in the nucleus (something that is reflective of its role as a transcriptional activator) in cells that undergo EMT (35).
Extracellular proteins.
Fibronectin is a high-molecular-weight glycoprotein that serves as a scaffold for fibrillar ECM (93). Because it is one of the first molecules to appear when the fibrillar ECM is formed, it has been used as an indicator of type 1 EMT associated with gastrulation, palate fusion, and neurulation (94). Even though fibronectin is an integral constituent of the fibrotic ECM associated with tissue fibrosis and the desmoplastic stroma in tumors, the utility of fibronectin as a type 2 and type 3 EMT biomarker is limited, in part, because it is produced by various cell types, including fibroblasts, mononuclear cells, and epithelial cells (95, 96). Both type 2 and type 3 EMT, however, are associated with increased fibronectin expression in vitro (67, 97).
Of the principal basement membrane constituents — type IV collagens, laminin, nidogen, and sulfated proteoglycans — that are downregulated during EMT, laminin is best established as a biomarker of the process. Laminins are heterotrimeric glycoproteins composed of one α chain, one β chain, and one γ chain (98). Currently, 15 different heterotrimers are known (98). With regard to EMT, most studies focus on laminin1 (α1β1γ1), which is present in the embryo from the peri-implantation period forward (99). Type 1 EMT during gastrulation and palate fusion is marked by loss of laminin1 (100). Both type 1 and type 2 EMT are associated with downregulation of laminin1 in vitro and disruption and loss of laminin1 in vivo (88, 101). By contrast, upregulation of laminin 5 (α3β3γ2) is associated with type 3 EMT in cancer and type 2 EMT in tissue fibrosis. Laminin5 is detected in the discontinuous laminin patterns associated with invasive cancers, and its expression is linked to type 3 EMT in breast carcinomas of the ductal type (102), hepatocellular carcinoma (103), and oral squamous carcinoma (104). Similarly, laminin5 is part of the fibrotic ECM associated with idiopathic pulmonary fibrosis (105). A correlation between laminin5 and EMT in the embryo has not yet been reported.
Transcription factors.
Fibroblast transcription site–1 (FTS-1) is a cis-acting regulatory element present in the promoter region of various EMT-associated genes, including those that encode FSP1, Twist, Snail1, high mobility group AT-hook 2 (HMGA2), LEF1, Ets-1, E-cadherin, β-catenin, ZO-1, α-SMA, and vimentin. CBF-A and KRAB-associated protein 1 (KAP-1) form a complex with FTS-1 to modulate gene activity. Formation of the CBF-A/KAP-1/FTS-1 complex induces type 2 EMT in kidney tubular epithelial cells (54). De novo expression of CBF-A, a member of the heterogeneous nuclear ribonucleoprotein A/B (hnRNP A/B) family, can be observed in type 2 and type 3 EMT associated with onset of fibrosis (54) and metastatic tumor formation (42). However, its expression is not confined to EMT, and it is involved in various other processes, including stimulation of Ig transcription (106). KAP-1 is also a ubiquitously expressed transcriptional corepressor or activator that is well known to bind KRAB domains on zinc finger proteins (106). In EMT, under the limited circumstances where it has been studied, KAP-1 seems to be an activator and, in the CBF-A/KAP-1/FTS-1 complex, a proximal master activator of EMT-associated genes (54).
The number of environmental factors that are known to induce EMT, including growth factors, ECM constituents, proteases, and hypoxia, is increasing. Despite the distinct nature of known stimulants, the EMT response is relatively uniform. This raises interest in potential key regulators of EMT that are at the intersection of various signaling pathways and control the EMT response. Snail transcription factors are one prominent example of a common downstream target of various signaling pathways that regulates EMT (61). Of the three vertebrate Snail family members of zinc finger proteins (Snail1, Snail2, and Snail3), the functionally equivalent Snail1 and Snail2 (which was formerly known as Slug) mediate EMT (61). In fact, all known EMT events during development, cancer, and fibrosis appear to be associated with Snail activation (61).
Snail is most widely recognized as a suppressor of E-cadherin expression, but it regulates various other aspects of the EMT phenotype, such as increased expression of mesenchymal cell/fibroblast markers (fibronectin and vitronectin), decreased expression of various epithelial markers (claudins, occludins, and cytokeratins), inhibition of proliferation through suppression of cyclin D proteins and cyclin-dependent kinase 4 (CDK4), increased MMP expression, and protection from cell death (through suppression of expression of caspases, DNA fragmentation factor, and Bcl-interacting death agonist). Snail transcriptional activity is regulated by control of its subcellular localization. Phosphorylation of Snail causes it to be exported from the nucleus into the cytoplasm, resulting in its inactivation as a transcription factor. In human renal biopsies, fibrosis is associated with increased Snail1 expression (107), and Snail activation in tamoxifen-inducible Snail1-transgenic mice results in type 2 EMT and renal fibrosis (107).
Twist is a basic helix-loop-helix protein that is transcriptionally active during lineage determination and cell differentiation. It is upregulated during early embryonic morphogenesis (108, 109), tissue fibrosis (54, 110), and cancer metastasis (63, 97, 111). In the development of metastatic cancer cells by type 3 EMT, Twist can act independently of Snail to repress E-cadherin (63) and to upregulate fibronectin and N-cadherin (97).
Forkhead box C2 (FOXC2) is another transcription factor that acts as a pleiotropic inducer of EMT. During embryonic development, FOXC2 is widely expressed and is required for angiogenesis, musculogenesis, and organogenesis of the kidney, heart, and urinary tract (112, 113). Postnatally, expression of FOXC2 is normally restricted to adipocytes (114). However, FOXC2 is also expressed in ductal breast cancers and metastatic breast cancer cell lines (115). Overexpression of any one of the EMT inducers TGF-β1, Snail, Twist, or Goosecoid increases FOXC2 expression, and overexpression of FOXC2 itself induces EMT, suggesting an important role for FOXC2 in type 3 EMT (115). A role for FOXC2 in EMT associated with embryogenesis or fibrosis has not been established yet.
Epigenetic EMT and microRNAs
By the time type 2 EMT finishes, there is a complete switch of cellular phenotype (4). There is a window, however, in which it can be reversed by pharmacological doses of BMP-7 (55). In epithelial cancers, genetic mutations typically accumulate to produce a primary tumor nodule. Since the act of metastasis often results in secondary tumor nodules, it is highly likely that metastatic cells generated by type 3 EMT are also reversible in the right tissue and under a new biologic context (19, 20). The hypothesis that epigenetic modifications mediate such bidirectional transitions (EMT and MET) raises the possibility of an “epigenetic EMT” as opposed to “genetic EMT”; the precise role of epigenetic modifications in type 3 EMT is only now receiving attention (116). In addition, two recent microRNA array analyses have identified microRNAs of the miR-200 family as markedly downregulated in TGF-β1–induced EMT and in cancer cell lines that displayed an EMT phenotype (117, 118). Both studies demonstrate that miR-200 microRNAs target the E-cadherin transcriptional repressors zinc finger E-box binding homeobox 1 (ZEB1) and ZEB2 (117, 118). Moreover, miR-200 microRNAs are downregulated in ductal and metaplastic breast cancers (117). Conversely, TGF-β1–induced EMT involving keratinocytes is associated with induction of miR-21 (119) and Twist-induced EMT involving breast cancer cells is associated with induction of miR-10b (120). The precise role of microRNAs in regulating all subtypes of EMT will undoubtedly emerge with more work.
Conclusions
We suggest that EMT can be parsed into three subtypes that all share a common mechanism. Somatic cells undergoing EMT are fated to move, and the newly acquired potential to migrate offers subtle evidence of a previous EMT event. When movement and cytoskeletal morphogenesis cannot be detected easily, changes in plasticity biomarkers of EMT are useful for the recognition of these phenotypic transitions. At the moment, the greatest promise for identifying this interesting phenomenon lies in analyzing a collection of markers — in effect, a collection of protein signatures. It is likely that all three subtypes of EMT will share part of a signature, but some aspects of the signature may be unique to a particular subtype. The most visible protein signatures include at least one changing cytoskeletal protein, transcription factor, adherence protein, and perhaps the changing levels of discrete microRNAs. It may come to pass that investigators will need to validate the presence of a common signature for EMT while characterizing the unique features of a particular subtype event. We hope that this approach may someday advance the possibility of identifying unique molecular instructions that will be good targets for subtype regulation.
Acknowledgments
E.G. Neilson is supported by NIH grant DK-46282, and M. Zeisberg is supported by NIH grants DK-074558 and DK-081687 and a grant from the Harvard Digestive Diseases Center. We regret that many other interesting findings could not be cited here due to space constraints; however, some of these are cited in other articles in this Review Series.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: CBF-A, CArG box–binding factor–A; DDR2, discoidin domain receptor tyrosine kinase 2; EMT, epithelial-mesenchymal transition; FOXC2, forkhead box C2; FSP1, fibroblast-specific protein 1; FTS-1, fibroblast transcription site–1; KAP-1, KRAB-associated protein 1; LEF, lymphoid enhancer–binding factor; MET, mesenchymal-epithelial transition.
Citation for this article: J. Clin. Invest. 119:1429–1437 (2009). 10.1172/JCI36183
References
Articles from The Journal of Clinical Investigation are provided here courtesy of American Society for Clinical Investigation
Full text links
Read article at publisher's site: https://doi.org/10.1172/jci36183
Read article for free, from open access legal sources, via Unpaywall:
http://www.jci.org/articles/view/36183/files/pdf

Free to read at www.jci.org
http://www.jci.org/cgi/content/full/119/6/1429
Free to read at www.jci.org
http://www.jci.org/cgi/reprint/119/6/1429.pdf
Free to read at www.jci.org
http://www.jci.org/cgi/content/abstract/119/6/1429
Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Article citations
Transcriptional responses to direct and indirect TGFB1 stimulation in cancerous and noncancerous mammary epithelial cells.
Cell Commun Signal, 22(1):522, 28 Oct 2024
Cited by: 0 articles | PMID: 39468555 | PMCID: PMC11514872
Allicin Ameliorated High-glucose Peritoneal Dialysis Solution-induced Peritoneal Fibrosis in Rats via the JAK2/STAT3 Signaling Pathway.
Cell Biochem Biophys, 25 Oct 2024
Cited by: 0 articles | PMID: 39448419
Chromosome 8q24 amplification associated with human hepatocellular carcinoma predicts MYC/ZEB1/MIZ1 transcriptional regulation.
Sci Rep, 14(1):24488, 18 Oct 2024
Cited by: 0 articles | PMID: 39424877 | PMCID: PMC11489779
β-Mangostin Alleviates Renal Tubulointerstitial Fibrosis via the TGF-β1/JNK Signaling Pathway.
Cells, 13(20):1701, 14 Oct 2024
Cited by: 0 articles | PMID: 39451219 | PMCID: PMC11505648
Efficacy of epidermal growth factor in suppressing inflammation and proliferation in pterygial fibroblasts through interactions with microenvironmental M1 macrophages.
Sci Rep, 14(1):22601, 30 Sep 2024
Cited by: 0 articles | PMID: 39349715 | PMCID: PMC11442942
Go to all (1,429) article citations
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
EMT and MET as paradigms for cell fate switching.
J Mol Cell Biol, 4(2):66-69, 02 Dec 2011
Cited by: 28 articles | PMID: 22140271
Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front.
Histochem Cell Biol, 130(3):481-494, 22 Jul 2008
Cited by: 145 articles | PMID: 18648847 | PMCID: PMC2522326
Review
 Free full text in Europe PMC
Free full text in Europe PMCTGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT).
Respir Res, 6:56, 09 Jun 2005
Cited by: 512 articles | PMID: 15946381 | PMCID: PMC1177991
Evidence for epithelial-mesenchymal transitions in adult liver cells.
Am J Physiol Gastrointest Liver Physiol, 291(4):G575-83, 18 May 2006
Cited by: 83 articles | PMID: 16710052
Funding
Funders who supported this work.
NIDDK NIH HHS (6)
Grant ID: DK-46282
Grant ID: R01 DK046282
Grant ID: DK-074558
Grant ID: DK-081687
Grant ID: K08 DK074558
Grant ID: R03 DK081687


