Efficient genome editing using tRNA promoter-driven CRISPR/Cas9 gRNA in Aspergillus niger.
![]() Free full text in Europe PMC
Free full text in Europe PMC
Abstract
Free full text
Efficient genome editing using tRNA promoter-driven CRISPR/Cas9 gRNA in Aspergillus niger
Abstract
As a powerful tool for fast and precise genome editing, the CRISPR/Cas9 system has been applied in filamentous fungi to improve the efficiency of genome alteration. However, the method of delivering guide RNA (gRNA) remains a bottleneck in performing CRISPR mutagenesis in Aspergillus species. Here we report a gRNA transcription driven by endogenous tRNA promoters which include a tRNA gene plus 100 base pairs of upstream sequence. Co-transformation of a cas9-expressing plasmid with a linear DNA coding for gRNA demonstrated that 36 of the 37 tRNA promoters tested were able to generate the intended mutation in A. niger. When gRNA and cas9 were expressed in a single extra-chromosomal plasmid, the efficiency of gene mutation was as high as 97%. Co-transformation with DNA template for homologous recombination, the CRISPR/Cas9 system resulted ~42% efficiency of gene replacement in a strain with a functioning non-homologous end joining machinery (kusA+), and an efficiency of >90% gene replacement in a kusA- background. Our results demonstrate that tRNA promoter-mediated gRNA expressions are reliable and efficient in genome editing in A. niger.
Introduction
The filamentous fungus Aspergillus niger is an important cell factory used in industry for the production of enzymes and organic acids. Owing to its genetic tractability, A. niger is widely used for research in fungal physiology, biochemistry and biotechnology. The availability of genome sequence of this organism [1,2] has facilitated numerous studies in gene function, gene regulation, primary and secondary metabolism [3,4]. The utility of A. niger as an industrial cell factory and as a model organism for research can be further improved by the development of a highly efficiency genome-editing system. https://www.ncbi.nlm.nih.gov/pubmed/17259976
Multiple nuclease-based gene targeting methods have been developed to improve the efficiency and precision of generating genetic modifications. These include Zinc-finger nuclease (ZFN) [5], Transcription Activator-Like Effector Nuclease (TALEN) [6], I-SceI meganuclease [7], and Clustered Regularly Interspaced Short Palindromic Repeat / associated nuclease 9 (CRISPR/Cas9) [8]. With the ability of nucleases to make DNA double strand break (DSB) in the host organism, nuclease-based gene targeting methods have been successfully used for gene disruption, knock-in mutation as well as improving heterologous protein production by integrating foreign genes to defined genomic loci [5,9,10]. The type II CRISPR/Cas9 system from Streptococcus pyogenes has revolutionized genome editing by providing a simple, flexible and efficient strategy to precisely edit the genomes of mammals [11], plant [12], fly [13], fungi [14] and bacteria [15]. Two components are essential in type II CRISPR/Cas9 system: a functional Cas9 nuclease and a chimeric guide RNA (gRNA) consisted of two regions, a CRISPR RNA (crRNA) harboring 20-nucleotide target-recognizing sequence at the 5’-end and a trans-activating crRNA (tracrRNA) for Cas9 binding. The crRNA is user-defined to match the target genomic locus. It guides the gRNA to form a RNA/DNA hybrid at the target genomic locus and recruits the Cas9 nuclease to generate DNA DSB [16]. Consequently, the CRISPR/Cas9 approach supports the editing of numerous genomic loci.
An efficient promoter that can facilitate gRNA transcription in vivo is a bottleneck of adoption of CRISPR/Cas9 system in Aspergilli. So far, in the reported CRISPR/Cas9 platform for A. niger, RNA polymerase II (Pol II) promoter and terminator were employed to drive the transcription of the gRNA, which required the inclusion of two self-splicing ribozyme sequences flanking the gRNA sequence to free the mature gRNA transcript [14]. The endogenous U6 promoter, an RNA polymerase III (Pol III) promoter, has been used for CRISPR/Cas9 genome editing in A. oryzae with an efficiency lower than 20% in generating gene mutations [17]. Another Pol III promoter, U3 promoter of A. fumigatus (AfU3), was successfully used to transcribe gRNA in A. nidulans. However, endogenous U6 and AfU6 promoters failed in generating functional gRNA in A. nidulans, which indicates that U6 promoters have limited function as gRNA promoters in fungi [18,19]. Therefore, it is important to find reliable promoters to drive gRNA transcription in vivo that can result in efficient genome editing of A. niger to expand its utility as a model and industrial organism.
In eukaryote cells, the transfer ribonucleic acid (tRNA) genes are transcribed by Pol III [20]. The transcription of fungal tRNA and other eukaryotes genes is constitutive and is independent of carbon source and cultivation conditions [21,22]. The tRNA promoter includes a region centered approximately 30 base pairs (bp) upstream of the tRNA gene start site and two intragenic sequences, Box A and Box B [20,23,24]. Their short sequences [25] and their self-splicing capacity [26,27] make tRNA promoters suitable for transcribing the short and non-coding gRNA sequence. The self-splicing ability of tRNA has been used to separate multiplexed gRNAs in plants [28] and recently in filamentous fungi [18]. To the best of our knowledge, using tRNA promoter to independently drive gRNA transcription has not been described in filamentous fungi. In this study, we describe the use of tRNA promoters and terminators to drive the gRNA transcription in vivo and show that this method is highly efficient in genome editing in A. niger.
Results
The use of tRNA promoters and terminators to drive gRNA transcription in A. niger
We manually curated the genome of the A. niger NRRL3 (http://gbrowse.fungalgenomics.ca/cgi-bin/gb2/gbrowse/Aspni_nrrl3_public) and identified 284 tRNA genes (chromosomal locations and sequences were listed in S1 Table). Six of the 284 tRNA genes are pseudogenes and three encode selenocystein tRNA. There are 8–24 tRNA genes for 17 of the 20 main amino acids. The three amino acids with fewer than eight corresponding tRNA genes are: tryptophan with one, asparagine with three, and cysteine with four tRNA genes (S1 Table). To determine the ability of tRNA promoters to drive the functional transcription of gRNA, we chose two random tRNA genes from every amino acid except that of asparagine and tryptophan. Three valine tRNAs were chosen due to the lack of intron in one and the difference in intron lengths in the two others. In total, 37 tRNA genes and their flanking sequences to use as promoters and terminators were selected (Table 1). Each gRNA cassette contains the following elements: 1) tRNA promoter which comprises the entire tRNA gene including introns, plus 100 nt upstream sequence; 2) the gRNA sequence that comprises a 20 nt crRNA guiding sequence that targets the albA gene (gene ID: NRRL3_00462) and a 80 nt tracrRNA sequence for Cas9 protein binding [29]; and 3) the 7–42 nt of tRNA terminator that spans from the end of tRNA gene to the poly-T stretch. The gene albA encodes a polyketide synthase involved in both dihydroxynaphthalene melanin and naphtho-gamma-pyrone synthesis that are required for spore pigmentation [30]. The disruption of albA prevents biosynthesis of black pigment, and results in white mutant conidia [14,30]. This phenotypic change provided a visual scoring of mutants directly on the transformation plate. For a quick, initial test of the ability of tRNA promoters and terminators to regulate gRNA transcription, we co-transformed the 37 linear gRNA cassettes independently into A. niger with cas9-bearing plasmid ANEp8-Cas9 (Fig 1A). The ANEp8-Cas9 plasmid was derived from the plasmid ANEp8 [31] that contains AMA1 sequence allowing extra-chromosomal replication [32] and the pyrG selection marker.

(A) The ANEp8-Cas9 plasmid used to express cas9 gene. (B) The construct of gRNA cassette and the ANEp8-Cas9-gRNA plasmid bearing both gRNA cassette and cas9 gene. The tRNA promoter is composed of a tRNA gene and 100 bp upstream sequences. Arrow indicates the start site of transcription. Abbreviations: bla, beta-lactamase gene conferring ampicillin resistance; pkiA Prom, promoter of the pyruvate kinase gene; glaA Term, terminator of the glucoamylase gene; NLS: nuclear localization signal.
Table 1
| tRNA promoter | Length of tRNA gene (bp) | Length of intron (bp) | Anticodon | Length of tRNA promoter (bp) | Length of tRNA terminator (bp) | Number of total colonies in transformation plates* | Number of white colonies (albA mutant) |
|---|---|---|---|---|---|---|---|
| tRNAAla5 | 72 | 0 | AGC | 172 | 16 | 88 | 9 |
| tRNAAla23 | 72 | 0 | AGC | 172 | 7 | 36 | 2 |
| tRNAArg8 | 72 | 0 | ACG | 172 | 9 | 96 | 12 |
| tRNAArg21 | 72 | 0 | ACG | 172 | 9 | 71 | 8 |
| tRNAAsp2 | 91 | 19 | GTC | 191 | 18 | 124 | 12 |
| tRNAAsp5 | 90 | 18 | GTC | 190 | 11 | 65 | 2 |
| tRNACys1 | 72 | 0 | GCA | 172 | 11 | 100 | 7 |
| tRNACys2 | 72 | 0 | GCA | 172 | 15 | 72 | 5 |
| tRNAGln1 | 72 | 0 | CTG | 172 | 16 | 72 | 6 |
| tRNAGln2 | 92 | 20 | TTG | 192 | 20 | 102 | 6 |
| tRNAGlu1 | 72 | 0 | TTC | 172 | 17 | 56 | 4 |
| tRNAGlu12 | 71 | 0 | TTC | 171 | 16 | 63 | 3 |
| tRNAGly5 | 71 | 0 | GCC | 171 | 26 | 68 | 0 |
| tRNAGly13 | 71 | 0 | GCC | 171 | 16 | 34 | 3 |
| tRNAHis2 | 84 | 12 | GTG | 184 | 42 | 62 | 4 |
| tRNAHis3 | 71 | 0 | GTG | 171 | 32 | 82 | 4 |
| tRNAIle4 | 74 | 0 | AAT | 174 | 16 | 60 | 3 |
| tRNAIle8 | 74 | 0 | AAT | 174 | 17 | 94 | 4 |
| tRNALeu6 | 111 | 27 | AAG | 211 | 17 | 148 | 8 |
| tRNALeu14 | 112 | 27 | AAG | 212 | 13 | 46 | 1 |
| tRNALys6 | 73 | 0 | CTT | 173 | 16 | 124 | 6 |
| tRNALys17 | 73 | 0 | CTT | 173 | 18 | 82 | 5 |
| tRNAMet4 | 72 | 0 | CAT | 172 | 15 | 84 | 2 |
| tRNAMet9 | 72 | 0 | CAT | 172 | 21 | 78 | 3 |
| tRNAPhe2 | 73 | 0 | GAA | 173 | 17 | 60 | 2 |
| tRNAPhe12 | 73 | 0 | GAA | 173 | 11 | 88 | 10 |
| tRNAPro1 | 91 | 19 | AGG | 191 | 17 | 57 | 7 |
| tRNAPro3 | 91 | 19 | AGG | 191 | 17 | 68 | 3 |
| tRNASer4 | 105 | 23 | GCT | 205 | 13 | 98 | 4 |
| tRNASer7 | 81 | 0 | AGA | 181 | 15 | 90 | 4 |
| tRNAThr5 | 73 | 0 | AGT | 173 | 15 | 46 | 4 |
| tRNAThr6 | 73 | 0 | AGT | 173 | 13 | 48 | 4 |
| tRNATyr3 | 88 | 13 | GTA | 188 | 22 | 63 | 6 |
| tRNATy7 | 88 | 13 | GTA | 188 | 21 | 42 | 2 |
| tRNAVal1 | 74 | 0 | CAC | 174 | 18 | 33 | 4 |
| tRNAVal4 | 96 | 23 | CAC | 196 | 25 | 45 | 1 |
| tRNAVal15 | 87 | 14 | TAC | 187 | 18 | 64 | 7 |
* The colonies number was from single transformations
In the control plate where the cells were transformed with ANEp8-Cas9 alone, only black colonies appeared (S1A Fig). Except in the case of tRNAGly5, all co-transformation yielded a mixture of black and white mutant colonies. These results suggest that almost all endogenous tRNA promoters and terminators are able to transcribe functional gRNA in the CRISPR/Cas9 system in A. niger. However, the co-transformation method resulted low inactivation rate of albA gene, ranging between 2 to 13% (Table 1). The relatively low rates of mutation using this co-transformation method is expected because the frequency of maintaining extra-chromosomal plasmid harboring the selectable marker is much higher than that of maintaining the integrated exogenous DNA [32]. In other words, only an uncontrolled subpopulation of transformants in these experiments is predicted to contain the linear gRNA cassette. Consequently, this initial experiment only reveals that most endogenous tRNA promoters and terminators are able to drive gRNA transcription and support CRISPR/Cas9 mutagenesis in A. niger, it does not provide quantitative information on the efficiency of different tRNA promoters in generating functional gRNA.
tRNA promoters are highly efficient in driving gRNA transcription
To compare the efficiency of different tRNA promoters in CRISPR/Cas9 system, the gRNA cassette was cloned into ANEp8-Cas9 to generate plasmid ANEp8-Cas9-gRNA (Fig 1B). We used extra-chromosomal plasmid for three reasons: 1) it supports high frequency of transformation [33]; 2) the transcription level of cas9 and gRNA are expected to be similar among transformants as they are not influenced by the site of integration; and 3) the plasmid can be removed easily by counter selection, leaving no exogenous DNA.
From the 36 positive tRNA-driven gRNAs (targeting albA) tested in the above co-transformation experiment, we chose eleven gRNA cassettes to construct plasmids containing both cas9 and gRNA. The eleven cassettes include tRNAs carrying hydrophilic (tRNAArg21, tRNACys1 and tRNAGln2), polar aliphatic (tRNAAsp5), aliphatic hydrophobic (tRNAAla5, tRNALeu6, tRNAPro1 tRNAVal1; 4; 15) and aromatic hydrophobic (tRNATyr3) amino acids. Among them three valine tRNA promoters were included because their tRNA genes contain either no intron or different intron lengths (Table 1).
The eleven ANEp8-Cas9-gRNA plasmids were transformed individually into A. niger strain N593 (Table 2). The results showed that ten out of eleven cassettes gave rise to transformants of which 82–96% were mutants (Table 3 and S1A Fig). The tRNAGln2 cassette resulted in significantly lower mutation rate of 15±6%. Additionally, the three tRNAVal promoters displayed a similarly high level of mutational efficiency, suggesting that tRNA introns do not influence its promoter capability in gRNA transcription.
Table 2
| A. niger strains | Genotype and/or Description | References |
| NRRL3 | Wild type | |
| N593 (ATCC 64973) | cspA pyrA6 | [34] |
| NRRL2270 (ATCC11414) | Citric acid producer | [35] |
| NRRL2270ΔpyrG | Uridine auxotroph | This study |
| NRRL2270ΔpyrGΔkusA | Uridine auxotroph and NHEJ deficient | This study |
| Plasmids | ||
| ANEp8-Cas9 | Extra-chromosomal cas9 expressing plasmid | This study |
| ANEp8-Cas9-gRNAalbA | Extra-chromosomal cas9 expressing plasmid with gRNA targeting albA locus | This study |
| ANEp8-Cas9-gRNAolvA | Extra-chromosomal cas9 expressing plasmid with gRNA targeting olvA locus | This study |
| ANEp8-Cas9-gRNAglaA | Extra-chromosomal cas9 expressing plasmid with gRNA targeting glaA locus | This study |
Table 3
| tRNA promoter | Gene disruption rate (%)* | |
|---|---|---|
| ΔalbA | ΔolvA | |
| tRNAAla5 | 93±2 | nd† |
| tRNAArg21 | 92±2 | 97±1 |
| tRNAAsp5 | 96±1 | nd |
| tRNACys1 | 82±6 | 96±2 |
| tRNAGln2 | 15±6 | 13±2 |
| tRNALeu6 | 82±7 | 95±2 |
| tRNAPro1 | 93±1 | 88±2 |
| tRNATyr3 | 82±11 | nd |
| tRNAVal15 | 82±14 | nd |
| tRNAVal4 | 90±3 | nd |
| tRNAVal1 | 94±1 | nd |
*The rate was calculated as the percentage of mutant colonies in all transformants. Results were based on triplicate transformations.
†nd: not determined.
To test the reliability of tRNA promoters in driving functional gRNA transcription in the CRISPR/Cas9 system, we targeted another gene olvA (ID: NRRL3_01039) that is also involved in the conidial pigmentation pathway. The loss of OlvA function results in olive-colored conidial mutants [30] (S1 Fig). We used five different tRNA promoters to drive gRNA to target olvA, four of which were randomly selected from the ten promoters that yielded >80% mutational frequency. We included the tRNAGln2 promoter to assess whether the low frequency of obtaining mutants is promoter dependent. The mutation rate of olvA was very similar to that observed towards albA (Table 3). A mutation rate of 13±2% was achieved by using tRNAGln2, while tRNAArg21, tRNACys1, tRNALeu6, and tRNAPro1 were able to induce mutation rates of between 88 to 97%. These results indicate that 1) the efficiency of gene editing by the CRISPR/Cas9 system is promoter-dependent; and 2) most endogenous tRNAs are very efficient and reliable in driving gRNA transcription in the CRISPR/Cas9 system.
Both deletions and insertions are generated by tRNA promoter-driven CRISPR/Cas9 mutagenesis
To determine the nature of CRISPR/Cas9-mediated mutations, we PCR-amplified the target genomic regions of the phenotypic mutants and sequenced the PCR products. Thirty transformants were analyzed, representing 15 albA and 15 olvA mutants mediated by five different tRNA promoters. These fragments covered the regions of albA and olvA that were expected to be cleaved by Cas9. Table 4 shows that deletions were the predominant mutation with 22 out of 30 of Cas9-induced mutations appeared as short deletions in the range of 1 to 27 bp at the Cas9 cleavage site. Additionally, five mutants contained long deletions of 612–1051 bp. Insertions were observed in three mutants. Sequence analysis indicated that the inserted sequences were derived from ANEp8-Cas9-gRNA plasmid. These observations suggest that double strand break mediated by Cas9 induces error-prone repair that resulted in mutation of the targeted gene in A. niger. The results also suggest that the extra-chromosomal plasmid can function as pseudo-donor template in the insertion of sequences.
Table 4
| tRNA promoter | Target Gene | Mutant number | Deletion* | Insertion | Mismatch |
|---|---|---|---|---|---|
| tRNAArg21 | albA | 1 | 6 bp (+1 - +6) | ||
| albA | 2 | 6 bp (+1 - +6) | |||
| albA | 3 | 4 bp (+1 - +4) | |||
| olvA | 1 | 8 bp (+1 - +8) | |||
| olvA | 2 | 612 bp (-11 - +429; +438 - + 609) | |||
| olvA | 3 | 72 bp (-1 - +1) | |||
| tRNACys1 | albA | 1 | 27 bp (-27 - -1) | ||
| albA | 2 | 854 bp (-854 - -1) | |||
| albA | 3 | 815 bp (-813 - +2) | 1 bp (T-817C) | ||
| olvA | 1 | 18 bp (+1 - +18) | |||
| olvA | 2 | 10 bp (-2 - +8) | |||
| olvA | 3 | 7 bp (-1 - +6) | |||
| tRNAGln2 | albA | 1 | 39 bp (-2 - -1) | ||
| albA | 2 | 12 bp (-1 - +11) | |||
| albA | 3 | 22 bp (-22 - -1) | |||
| olvA | 1 | 13 bp (-12 - +1) | |||
| olvA | 2 | 1096 bp (-1038 - +58) | |||
| olvA | 3 | 11 bp (-8 - +3) | |||
| tRNALeu6 | albA | 1 | 1 bp (-1) | ||
| albA | 2 | 15 bp (-3 - +12) | |||
| albA | 3 | 979 bp (-979 - -1) | |||
| olvA | 1 | 1 bp (-1) | |||
| olvA | 2 | 88 bp (-1 - +4) | |||
| olvA | 3 | 13 bp (-13 - -1) | |||
| tRNAPro1 | albA | 1 | 6 bp (-7 - -2) | ||
| albA | 2 | 4 bp (-3 - +1) | 1 bp (A-6G) | ||
| albA | 3 | 2 bp (-2 - -1) | |||
| olvA | 1 | 1 bp (-1) | |||
| olvA | 2 | 2 bp (-2 - -1) | |||
| olvA | 3 | 1 bp (-1) |
*The mutation locations are given in brackets. The Cas9 cleavage site is defined as 3 bp upstream of the PAM sequence and is designated position "0". The negative and positive coordinates indicate mutations locate at 5’ and 3’ to the position 0, respectively.
CRISPR/Cas9 mediates gene replacement in A. niger kusA+ and kusA- backgrounds with different efficiencies
To evaluate how CRISPR/Cas9-mediated DSB can improve gene targeted replacement in A. niger, we constructed a ANEp8-Cas9-gRNAglaA plasmid targeting the glaA locus, which is a highly expressed gene encoding glucoamylase, and a linear gene replacement cassette as donor template (S2 Fig). The linear donor template comprised the coding region of adaR (gene ID: NRRL3_09545) flanked by 600 bp of glaA promoter and terminator sequences. The adaR gene encodes a transcription regulator involved in the biosynthesis of TAN-1612/BMS-192548, a pigmented polyketide secondary metabolite [36]. The integration and overexpression of adaR yields orange colonies because of the overproduction of TAN-1612/BMS-192548 [36]. Orange colonies can be directly counted on transformation plates. As a control, we transformed donor DNA with the plasmid ANEp8-Cas9 without the gRNA cassette. In addition, we created the kusA- mutant strain according to Meyer et al. [35] and described in S3 and S4 Figs to evaluate the role of non-homologous end joining (NHEJ) DNA repair in mediating CRISPR/Cas9 mutagenesis. Mutants deficient in KusA are defective in NHEJ and cannot repair DNA DSBs [37,38]. Previous studies showed improved homologous recombination in NHEJ-deficient A. niger strains [38–40]. However, little is known about the efficiency of homologous recombination when DSB is mediated by CRISPR/Cas9.
In order to easily distinguish gene integration mutants from abortive or young transformants, nine orange colonies from each transformation experiment were chosen for PCR screening using primers outside the glaA locus (S2 Fig). Table 5 shows that in the kusA+ background, without the gRNA no gene replacement occurred. In the presence of the gRNA in the kusA+ background, about 75% of the transformants displayed orange colonies and 5 of 9 orange colonies tested were the results of homologous recombination, suggesting a frequency of gene replacement of about 42%. In the kusA- background, the absence of gRNA yielded 21 orange colonies out of 75 transformants with 6 of 9 orange colonies tested positive for homologous recombination, suggesting a frequency of gene replacement of about 20%. This finding supports previous observation that the turning off of NHEJ repair that improves homologous recombination in A. niger [39,40]. In the kusA- background and in the presence of gRNA, orange colonies comprised about 93% of the transformants with all 9 orange colonies tested positive for homologous recombination. This would imply a gene replacement rate of ~93%. In addition, in the kusA- strain, no transformant was found in the experiment in which only the ANEp8-Cas9-gRNAglaA plasmid was transformed because fungal cells could not repair the DSB in the absence of a repair donor template (Table 5). Based on this reasoning, we would expect 100%, instead of 93%, gene replacement frequency in the situation where we have functional gRNA and a gene replacement DNA template in the kusA- background. The few non-orange transformants could be the result of less than 100% efficiency of the gRNA and/or some of the young transformants scored did not have sufficient time to manifest the mutant phenotype. Since kusA- strains cannot repair DSB, constructing mutants in the kusA- genetic background using CRISPR/Cas9 and rescue templates has the advantage that off-target mutations would be minimized as ectopic mutations would result in unrepaired chromosomal breaks and cell lethality.
Table 5
| CRISPR plasmid | adaR donor template | NHEJ competent strain (kusA+) | NHEJ deficient strain (kusA-) | ||||
|---|---|---|---|---|---|---|---|
| Total colonies* | Orange colonies*† | Gene replacement mutants / Screened mutants‡ | Total colonies* | Orange colonies*† | Gene replacement mutants / Screened mutants‡ | ||
| ANEp8-Cas9 | Yes | 125 ± 3 | 15 ± 1 | 0/9 | 75 ± 1 | 21 ± 5 | 6/9 |
| ANEp8-Cas9-gRNAglaA | Yes | 106 ± 4 | 80 ± 3 | 5/9 | 88 ± 7 | 82 ± 6 | 9/9 |
| ANEp8-Cas9-gRNAglaA | No | 98 ± 6 | - | - | 0 | - | - |
*The value was calculated from duplicate independent transformations and the average is mentioned.
†The mutant number was counted according to the phenotype of orange color in the primary transformants.
‡From each scenario, nine of the orange colored transformants were selected to screen for gene replacement by PCR.
Discussion
Besides an active Cas9 protein, a functional gRNA is an essential component in CRISPR/Cas9 system to engineer the host genome. For delivering gRNA in vivo, the formation of functional gRNA requires an efficient promoter to drive gRNA transcription and a splicing mechanism to form mature gRNA transcript. In this study, we describe a strategy using Pol III tRNA promoters to drive gRNA transcription. This study shows that the self-splicing ability of tRNAs is highly efficient in generating functional gRNAs.
Since 2015, the CRISPR/Cas9 system in genome editing has been established in several genera of filamentous fungi. As summarized in Table 6, the promoter selection has shifted from Pol II promoter (e.g. pgdA and trpC promoters) to Pol III promoters for the in vivo transcription of gRNA. This is because Pol III promoter is more suitable for in vivo transcription of short non-coding transcripts such as gRNA [20,41]. In addition, the short length of Pol III promoters and terminators makes it easier to construct gRNA cassette. Among Pol III promoters used in filamentous fungi CRISPR/Cas9 platforms, the species-specific U6 promoter is the most widely used [17,42–47], and the SNR52 promoter from Saccharomyces cerevisiae has been tested in Neurospora crassa [48] and A. fumigatus [49] (Table 6). In this study, the tRNA promoters contain the entire sequence of tRNA gene plus 100 nt upstream sequence with a total sequence varying from 171 to 272 nt (S2 Table), which is similar as the lengths of U6 (200 to 250 nt) and S. cerevisiae SNR52 (269 nt) promoters [29,50]. The main challenge of current Pol III gRNA promoters such as U3 and U6 is that the same type of promoter results in highly variable gene editing efficiency amongst different species. Unlike U6 promoter that is an upstream element outside of the transcription start site [50], the tRNA gene is transcribed together with gRNA sequence [20,51]. In the cell the tRNA transcript is posttranscriptionally cleaved at the 3‘-end by RNase Z [27,28]. Accordingly, tRNA promoter regulated system is able to release mature functional gRNA transcript as demonstrated in this study and elsewhere [52].
Table 6
| Organism | gRNA cassette components | Gene replacement rate (%) | Gene disruption rate (%) | Expression plasmid/other method | References | ||
|---|---|---|---|---|---|---|---|
| Promoter | Ribozyme splicing sequences | Terminator | |||||
| A. niger | tRNA | No | tRNA | 42% or 93%* | 13–15% or 82–97%† | Extrachromosomal (AMA1) | This study |
| A. nidulans | gpdA | Yes | trpC | 90% | 20–30% | Extrachromosomal (AMA1) | [14] |
| A. aculeatus | gpdA | Yes | trpC | 65% | > 70% | Extrachromosomal (AMA1) | [14] |
| A. fumigatus | snr52 | No | sup4 | nd‡ | 25–53% | Integrative | [49] |
| A. fumigatus | U6-1/2/3 | No | U6 | 63% | 43% | Extrachromosomal (AMA1) | [43] |
| A. oryzae | U6 | No | U6 | nd | 10–20% | Integrative | [17] |
| N. crassa | snr52 | No | sup4 | nd | nd | Integrative | [48] |
| Pyricularia oryzae | U6-1/2 trpC | No Yes | U6 trpC | 36–84% 10–27% | nd nd | Integrative | [42] |
| T. reesei | In vitro transcription | 93–100% | nd | in vitro | [53] | ||
| Ustilago maydis | U6 | No | U6 | nd | 70–100% | Extrachromosomal (ARS) | [44] |
| Penicillium chrysogenum | U6 tRNA | No No | U6 tRNA | 100% nd | nd nd | Extrachromosomal (AMA1) | [45] |
| A. carbonarius | gpdA | Yes | trpC | 100% | nd | Extrachromosomal (AMA1) | [54] |
| Coprinopsis cinerea | U6 | No | U6 | nd | 21% | Integrative | [47] |
| Myceliophthora thermophila | U6 | No | U6 | 95% | nd | Integrative | [46] |
| M. heterothallica | U6 | No | U6 | 90% | nd | Integrative | [46] |
*The value of 42% and 93% were obtained from experiments in A. niger kusA- and kusA+ strains respectively using tRNAPro1 as gRNA cassette promoter.
†The 13–15% correspond to tRNAGln2 promoter, and the 82–97% correspond to the other ten tRNA promoters as described in this study.
‡nd: not determined.
By introducing upstream sequences, the tRNA promoters in our constructs include all external and internal binding sites of Pol III transcription factors needed for transcription to proceed normally. Except tRNAGln2, the tRNA promoters tested in this study lead to high rate of gene mutation (82–97%). In endogenous U6 promoter-driven platforms, the mutation rate is highly variable among different fungal species and ranges from 10 to 100% (Table 6). In a recent study, the endogenous U6 promoter and the heterologous AfU6 promoter failed to transcribe gRNA in CRISPR/Cas9 system in A. nidulans [18]. In addition, gRNAs transcribed by S. cerevisiae SNR52 promoter resulted in mutation rates of between 25 to 53% in A. fumigatus [49] (Table 6). These studies show that the type of promoters used to transcribe gRNA has a high impact in CRISPR/Cas9 activity. Overall results show that expressing Cas9 and the gRNA in single plasmid improve gene deletion and gene replacement. In addition no difference on growth was observed when transformed A. niger with ANEp8-Cas9 or ANEp8-Cas9-gRNA in the present study, except the color phenotype (S1A and S1B Fig). This study provides a broad list of highly efficient tRNA promoters for gRNA transcription.
In A. niger and other eukaryotes, DSB created in the genome is repaired by NHEJ or by homologous recombination [55]. The NHEJ is the most dominant pathway in A. niger and the frequency of homologous recombination increases with the deletion of the kusA gene, which is involved in the NHEJ repair mechanism [56]. In this study, we tested CRISPR/Cas9 mediated gene replacement by homologous recombination in both kusA- and kusA+ strains. By targeting the glaA locus, we demonstrated that the CRISPR/Cas9 method can increase the frequency of gene replacement in A. niger (Table 5). In gene replacement mediated by CRISPR/Cas9 activity, our results are comparable to the results obtained in filamentous fungi such as A. nidulans, Trichoderma reesei and Myceliophthora thermophila, and show higher frequency comparing to findings in A. aculeatus, A. fumigatus, Penicillium chrysogenum and A. carbonarius (Table 6). In summary, the tRNA promoter-driven gRNA transcription system is very efficient to generate DNA double-strand break for gene mutation mediated by NHEJ and for gene replacement by homologous recombination in kusA+ and kusA- strains. This powerful tool will facilitate further improvement of the industrial fungus A. niger.
Material and methods
CRISPR design
tRNA gene prediction and selection
The tRNA gene and promoter sequences for this study were retrieved from the genome sequence of A. niger NRRL3 (data not published, available at http://genome.fungalgenomics.ca). We used four tRNA gene electronic tools tRNAscan-SE version 1.3 [57], SPLITSX [58], ARAGORN [59] and tRNAfinder [60] to predict all potential cytoplasmic tRNA genes in the nuclear genome of A. niger NRRL3. A total of 451 tRNA gene models were predicted by the four tools. We manually reviewed the predicted models to ensure that they conform to the length and structure of tRNAs and that the terminator contains a stretch of consecutive thymidines (poly-T) [61,62]. Where multiple non-identical models are detected in the same genomic region, we chose the consensus model predicted by more than one program. Since no fungal tRNA gene has been found to have intron exceeding 100 nucleotides [63], we removed 14 tRNA models predicted by ARAGORN to have introns >100 nucleotides. The final set contains 284 tRNA gene models (S1 Table).
Identification of guide sequences
We used Geneious R9.1 software [64] to search 20 bp guide sequences against the A. niger NRRL3 genome. S3 Table shows the guide sequences used in this study, and none of them possesses off-target potential.
Construction of gRNA cassette
The gRNA cassettes were assembled by fusion PCR of two DNA fragments, and the primers used were listed in S4 Table. The first fragment was the tRNA promoter that was amplified from A. niger genomic DNA and the 20 bp guide sequence was added at the 3’-end through PCR using as overlapping sequence. The second fragment was amplified from the synthesized 80 bp tracRNA template [29] with the 20 bp guide and tRNA terminator sequences (S3 Table) added at 5‘and 3‘ends respectively via PCR. In the fusion PCR, 0.5μL of each unpurified PCR fragments were used as template in a 100 μL reaction and performed with Phusion DNA polymerase (Thermo Scientific).
Plasmid construction
Escherichia coli DH5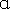 was used as the host strain for maintenance and production of plasmid DNA.
was used as the host strain for maintenance and production of plasmid DNA.
Construction of plasmid ANEp8-Cas9
The coding sequence of S. pyogenes cas9 (Uniprot Q99ZW2) was codon optimized for translation in A. niger. A nucleotide sequence encoding a nuclear localization signal (NLS) peptide SRADPKKKRKV was fused to the carboxy terminus of cas9. The DNA sequence of Cas9-NLS was synthesized by IDT Inc. (IA, USA), and amplified by primers Cas9-F and Cas9-R (S4 and S5 Tables) to add PacI and FseI restriction sites at the 5’ and 3’ ends, respectively. Restriction enzyme digests by PacI and FseI were used to clone cas9-NLS downstream of the pkiA promoter in the expression vector ANEp8 [31]. The resulting cas9-containing plasmid was designated as ANEp8-Cas9 (S5 Table).
Construction of plasmid ANEp8-Cas9-gRNA
A 38-bp ligation independent cloning (LIC) site centered with SwaI restriction site (S5 Fig) was introduced into ANEp8-Cas9 plasmid via PCR amplification. The resulting plasmid was used as host vector to harbor the gRNA cassette by using the LIC method [65]. The gRNA cassette used for plasmid construction was amplified with a pair of end primers to link with LIC sequence sites at the both sides as illustrated in S5 Fig, to generate complementary single-strand overhangs between ANEp8-Cas9 vector and gRNA cassette insert. The linearized ANEp8-Cas9 and the gRNA cassette DNA (ending with LIC tails) were treated by T4 DNA polymerase in the presence of dGTP and dCTP, respectively. The 20 μL reaction mixture contained 0.2 pmol of DNA, 0.8 μL of 100 mM dithiothreitol, 2 μL of 25 mM dGTP or dCTP, and 3 U of T4 DNA polymerase in NEB buffer 2.1. The reaction was carried out at 22°C for 30 minutes followed by enzyme inactivation by heating at 75°C for 20 minutes. The insert and vector were mixed in a 3:1 molar ratio. To achieve annealing, the mixture was first heated at 60°C for 5 minutes and then gradually decreased to 4°C (reduce 0.1°C per second). The annealed products were transformed into E.coli DH5α competent cells to generate plasmid ANEp8-Cas9-gRNA. All plasmids are available upon request.
Construction of adaR cassette for gene replacement at the glaA locus
The linear adaR (gene ID: NRRL3_09545) cassette with glaA flanking regions was constructed as shown in S2 Fig and S5 Table. Using genomic DNA of the A. niger strain NRRL2270 as template, 600 bp of 5’and 3’ regions flanking the coding region of the glaA gene as well as the adaR gene were amplified by primers with complementary ends (S4 Table). Based on their terminal overlaps, the three fragments were joined through fusion PCR amplification, resulting in an adaR cassette that was used for gene replacement at the glaA locus. A total of 5 μg of purified linear adaR cassettes were used in transformation. The PCR screening for targeted integration was carried out by using primers Fw_glaA-ext and Rv_glaA-ext (S2 Fig and S4 Table).
CRISPR/Cas9 genome editing in A. niger
Strain, media and growth conditions
Table 2 lists the A. niger strains used in this study. The uridine auxotrophic A. niger strain N593 was used for gene editing and to test tRNA promoter efficiency by CRISPR/Cas9. The strains NRRL2270ΔpyrG and NRRL2270ΔpyrGΔkusA were used for testing gene replacement by CRISPR/Cas9. All A. niger strains were grown on minimal medium [66] or on complete medium (minimal medium supplemented with 5 g/L of yeast extract and 1 g/L of casamino acids). As required, a final concentration of 1.3 mg/mL of 5-fluoroorotic acid (5-FOA) and/or 10 mM of uridine were added. The liquid media were supplemented with 15% D-maltose. The transformants were cultured at 30°C for 4 days.
Transformation, mutant purification and screening
Transformation and genomic DNA isolation of A. niger strains were performed as described previously [3]. All transformations were carried out using 1 μg of plasmid DNA together with 5 μg of linear cassette DNA when needed. Triplicate transformations were performed for each tRNA promoter tested, and duplicate transformations were carried out for the gene replacement of glaA. Mutant clones were visually identified according to conidia color. The CRISPR efficiency was calculated by dividing the number of mutant colonies by the total number of transformants. Three mutant colonies were randomly selected from the transformation plate and streak-purified twice on new minimal medium plates without uridine. Mutation patterns were analyzed by PCR amplification of albA (1.8 kb) and olvA (2.2 kb) sequences surrounding the CRISPR/Cas9 cut site using primers listed in S4 Table. Amplified DNA bands were visualized on agarose gel. Detailed information on the mutations was determined by DNA sequencing of the PCR products.
Supporting information
S1 Fig
An example of transformation plates for gene mutations and growth of purified transformants.A) From left to right are the transformation plates of A. niger N593 cells transformed with ANEp8-Cas9 plasmid (only expressed cas9), and ANEp8-Cas9-gRNA plasmid bearing tRNAPro1-driven gRNA to disrupt albA (98% efficiency) and olvA (95% efficiency) respectively. B) Growth phenotype of purified colonies of A. niger transformed with ANEp8-Cas9 and ANEp8-Cas9-gRNAolvA.
(PDF)
S2 Fig
Gene replacement of glaA gene.(A) Schematic illustration of targeted replacement by homologous recombination. (B) PCR screening for CRISPR/Cas9-mediated homologous integration among the nine-selected orange transformants from kusA+ (left) and kusA- (right) strains. The PCR was performed using their genomic DNA as template with primers outside of glaA locus. Replacement of glaA by adaR gave a 4.2 kb PCR fragment. Random integration of adaR cassette would leave the glaA locus intact, resulting in a 3.8 kb PCR fragment.
(PDF)
S3 Fig
Construction of kusA deletion cassette and strategy to generate NRRL2270ΔkusAΔpyrG strain.(A) Fusions PCR to construct the kusA deletion cassette by including 500 bps repeat of the 3’ UTR in 5’ of the pyrG selection marker to facilitate the loop out of the selection marker. (B) Loop out strategy of the pyrG selection marker to generate ΔkusAΔpyrG strain for subsequent transformation with the same selection marker: (1) Homologous integration of the kusA deletion cassette to remove the kusA gene in A. niger; (2) Generation of ΔkusAΔpyrG genotype in presence of 5-FOA plus uridine to facilitate pyrG loop out (3).
(PDF)
S4 Fig
PCR screening of transformants of ΔkusApyrG+ and ΔkusAΔpyrG genotypes respectively and the growth comparison with parental strain.(A) Screening of transformants for ΔkusApyrG+ genotype with primers binding externally to the kusA locus and primers binding within the pyrG selection marker. Two PCR bands with size of 3 kb and 2.5 kb respectively are expected by using primer pairs P1_ext/P6_int and P5_int/P8_ext. Transformants 3, 6, 9 and 13 have the deletion cassette integrated in the kusA locus and have ΔkusApyrG+ genotype, when transformants 4, 5, 7, 8, 10, 11, 12 and 14 have partial integration of the deletion cassette. For transformants lacking pyrG, the primers are not able to bind and amplify the gDNA of the parental strain (PS) NRRL2270pyrG-. (B) Screening for transformants with pyrG loop out. Spores of transformant 3 (NRRL2270ΔkusApyrG+) have been plated on minimal medium containing 5-FOA and uridine for 5 days. Colonies growing on 5-FOA are selected for screening pyrG selection marker loop out phenotype by PCR. A correct loop out of the pyrG marker is characterized by a PCR giving a band size of 2.2 kb, while a strain failed in pyrG loop out will display a band of 4 kb. Transformant 3’ with the correct loop out of the pyrG selection marker is NRRL2270ΔkusAΔpyrG and has been selected for further work. (C) Growth comparison of NRRL2270ΔpyrG and NRRL2270ΔkusAΔpyrG. One million spores of strains NRRL2270ΔkusAΔpyrG and NRRL2270ΔpyrG were plated on the uridine-contained minimal or complete medium, and incubated at 30°C for 3 days.
(PDF)
S5 Fig
The construction of ANEp8-Cas9-gRNA plasmid using LIC method.In ANEp8-Cas9 linearized vector, the single-strand 5' overhangs for LIC cloning were achieved by 3' →5' exonuclease activity of T4 DNA polymerase in the presence of dGTP. For gRNA cassette, the LIC tails were added to the ends via PCR, and the reverse complementing overhangs were generated by the same T4 DNA polymerase treatment process with dCTP. Through base pairing, plasmid ANEp8-Cas9-gRNA was assembled by annealing the complementary sequences between gRNA insert and plasmid vector. The complementary LIC sequences in the gRNA insert and ANEp8-Cas9 vector are shown in the figure.
(PDF)
S1 Table
The 284 predicted tRNA genes from A. niger NRRL3 genome.(XLSX)
S2 Table
DNA sequence of the 37 tRNA promoters and terminators used in the functional test.(XLSX)
S3 Table
The guide sequences used in this study.(DOCX)
S5 Table
DNA sequences of plasmids ANEp8-Cas9 and ANEp8-Cas9-gRNAglaA, and linear construct of adaR.(PDF)
Funding Statement
This work was supported by NSERC Strategic Industrial Biocatalysis Network. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data Availability
All gene sequences from A. niger and the genome sequence of A. niger NRRL3 are available at http://gbrowse.fungalgenomics.ca/cgi-bin/gb2/gbrowse/Aspni_nrrl3_public.
References
Articles from PLOS ONE are provided here courtesy of PLOS
Full text links
Read article at publisher's site: https://doi.org/10.1371/journal.pone.0202868
Read article for free, from open access legal sources, via Unpaywall:
https://journals.plos.org/plosone/article/file?id=10.1371/journal.pone.0202868&type=printable

Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Smart citations by scite.ai
Explore citation contexts and check if this article has been
supported or disputed.
https://scite.ai/reports/10.1371/journal.pone.0202868
Article citations
ReaL-MGE is a tool for enhanced multiplex genome engineering and application to malonyl-CoA anabolism.
Nat Commun, 15(1):9790, 12 Nov 2024
Cited by: 0 articles | PMID: 39532871 | PMCID: PMC11557832
Competition between homologous chromosomal DNA and exogenous donor DNA to repair CRISPR/Cas9-induced double-strand breaks in Aspergillus niger.
Fungal Biol Biotechnol, 11(1):15, 15 Oct 2024
Cited by: 0 articles | PMID: 39407321 | PMCID: PMC11481784
NHEJ and HDR can occur simultaneously during gene integration into the genome of Aspergillus niger.
Fungal Biol Biotechnol, 11(1):10, 05 Aug 2024
Cited by: 0 articles | PMID: 39103967 | PMCID: PMC11301975
Increasing the efficiency of CRISPR/Cas9-mediated genome editing in the citrus postharvest pathogen Penicillium digitatum.
Fungal Biol Biotechnol, 11(1):8, 13 Jul 2024
Cited by: 0 articles | PMID: 39003486 | PMCID: PMC11245846
Utilization of CRISPR-Cas genome editing technology in filamentous fungi: function and advancement potentiality.
Front Microbiol, 15:1375120, 28 Mar 2024
Cited by: 0 articles | PMID: 38605715 | PMCID: PMC11007153
Review
 Free full text in Europe PMC
Free full text in Europe PMC
Go to all (53) article citations
Data
Data behind the article
This data has been text mined from the article, or deposited into data resources.
BioStudies: supplemental material and supporting data
Genes & Proteins
- (1 citation) UniProt - Q99ZW2
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
Visualized Multigene Editing System for Aspergillus niger.
ACS Synth Biol, 10(10):2607-2616, 24 Sep 2021
Cited by: 7 articles | PMID: 34555894
A simple approach to mediate genome editing in the filamentous fungus Trichoderma reesei by CRISPR/Cas9-coupled in vivo gRNA transcription.
Biotechnol Lett, 42(7):1203-1210, 16 Apr 2020
Cited by: 8 articles | PMID: 32300998
Development of a gRNA Expression and Processing Platform for Efficient CRISPR-Cas9-Based Gene Editing and Gene Silencing in Candida tropicalis.
Microbiol Spectr, 10(3):e0005922, 11 May 2022
Cited by: 0 articles | PMID: 35543560 | PMCID: PMC9241840
Optimization of genome editing through CRISPR-Cas9 engineering.
Bioengineered, 7(3):166-174, 01 Apr 2016
Cited by: 37 articles | PMID: 27340770 | PMCID: PMC4927198
Review
Free full text in Europe PMC
Funding
Funders who supported this work.


