A review of current developments in RNA modifications in lung cancer.
Review![]() Free full text in Europe PMC
Free full text in Europe PMC
Abstract
Free full text
A review of current developments in RNA modifications in lung cancer
 1,2 and
1,2 and Abstract
Lung cancer has the highest incidence and mortality rates worldwide and is the primary cause of cancer-related death. Despite the rapid development of diagnostic methods and targeted drugs in recent years, many lung cancer patients do not benefit from effective therapies. The emergence of drug resistance has led to a reduction in the therapeutic effectiveness of targeted drugs, highlighting a crucial need to explore novel therapeutic targets. Many studies have found that epigenetic plays an important role in the occurrence of lung cancer. This review describes the biological function of epigenetic RNA modifications, such as m6A, m5C, m7G, and m1A, and recent advancements in their role in the development, progression, and prognosis of lung cancer. This review aims to provide new guidance for the treatment of lung cancer.
Introduction
Lung cancer, due to its low survival rate and high postoperative recurrence rate, is the primary cause of cancer-related death [1]. Lung cancer can be histologically divided into two groups, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [2]. NSCLC is the dominant histopathological subtype of lung cancer, accounting for 85% of all lung cancer cases, and can be further subdivided into lung squamous cell carcinoma (LUSC), lung adenocarcinoma (LUAD), or large cell carcinoma (LCC), with LUAD accounting for approximately 60% of all NSCLC cases [3]. NSCLC has a high mortality rate and poor prognosis due to its high malignancy, susceptibility to drug resistance, and high invasiveness. Despite the rapid development of diagnostic methods and targeted drugs in recent years, many lung cancer patients do not benefit from effective targeted therapies. The emergence of drug resistance in a large proportion of patients has led to a reduction in the therapeutic efficacy of targeted drugs [4]. The role of epigenetic modifications has only recently been implicated in the initiation and progression of various tumors. Epigenetic modifications refer to reversible changes in gene expression without involving the underlying nucleic acid sequence, thereby altering information processing. Key modifications include histone modification, DNA methylation, and RNA modification [5]. Although epigenetic modification and links to cancer have been extensively studied, the current findings do not fully elucidate the molecular mechanisms. The purpose of this review is to discuss the biological function and recent advances in the role of RNA modifications in lung cancer development, progression, and metastasis.
RNA modifications
There is growing evidence that different post-transcriptional RNA modifications exist across almost all organisms [6]. RNA modification has been identified as one of the important components of epigenetic regulation and pathological processes, particularly cancer, by modulating the behavior and biological function of RNA at the post-transcriptional level. At present, more than 170 different chemical modifications have been reported in RNA, including acetylation, phosphorylation, adenylylation, and methylation. Methylation is the most common modification, which includes N6-methyladenosine (m6A), 5-methylcytidine (m5C), N7-methylguanosine (m7G), and N1-methyladenosine (m1A). While drugs targeting histone modification and DNA modification have entered the clinical application stage, research on therapeutic strategies targeting RNA epigenetic modification remains in the early stages.
m6A RNA methylation
m6A is the most abundant and widely studied post-transcriptional RNA modification and refers to methylation at the sixth position of the adenylate nucleotide [7]. As early as the 1970s, m6A was recognized as a major modification in eukaryotic cell messenger RNA (mRNA) and other non-coding RNA. m6A has demonstrated regulatory roles in protein production by influencing mRNA stability, translation, splicing, and processing, as well as embryonic development and cell differentiation [8, 9]. The insertion of m6A is a dynamically reversible process, and relies on three types of methylation regulators: writers (methyltransferases), erasers (demethylases), and readers (binding proteins), collectively termed the writing-erasing-reading (WER) system (summarized in Table 1). Writers catalyze the installation of m6A and consist of methyltransferase-like protein 3 (METTL3), METTL14, and their accessory proteins RNA-binding motif protein 15 (RBM15), zinc finger CCCH-type containing 13 (ZC3H13), Wilms Tumor 1 associated protein (WTAP), and KIAA1429. Erasers remove the m6A modifications by demethylation and include AlkB homolog 5 (ALKBH5) and FTO-alpha-ketoglutarate-dependent dioxygenase (FTO) [10, 11]. Readers recognize m6A modifications on RNA and include insulin-like growth factor 2 mRNA binding proteins (IGF2BP), heterogeneous nuclear ribonucleoprotein (hnRNP), and members of the YTH family [12, 13]. The IGF2BP family consists of four distinct C-terminal KH domains, responsible for identifying m6A modifications, and two N-terminal RNA recognition motifs (RRMs) [14], that together promote the stability and translation of mRNA downstream target genes [15]. IGF2BP3, a member of the IGF2BP family, functions as a transcriptional regulator with implications for proliferation, invasion, and chemotherapy resistance of tumor cells [16, 17]. Elevated expression of IGF2BP3 has been consistently associated with poorer prognosis in lung cancer patients [18]. At present, evidence suggests that m6A modification regulates a series of malignant biological behaviors, and targeting m6A and associated regulatory proteins may be a promising method to prevent tumor progression [19]. However, more studies are needed to fully elucidate the complex mechanism of m6A modification in the regulation of the initiation and development of cancer.
Table 1
Regulatory proteins of RNA modification
| Modification | Structure | Writer | Eraser | Reader |
|---|---|---|---|---|
| m6A |  | RBM15 ZC3H13 METTL3,14 WTAP KIAA1429 | ALKBH5 FTO | IGF2BP hnRNP YTHDF1-3 YTHDC1-2 |
| m5C | 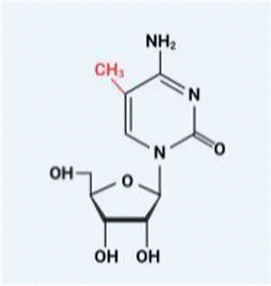 | DNMT2 NSUN1-7 | TET2 | ALYREF YBX1 |
| m7G | 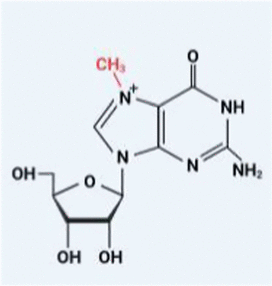 | METTL1 WDR4 | – | – |
| m1A |  | TRMT6 TRMT61A TRMT61B TRMT10C NML | FTO ALKBH1 ALKBH3 ALKBH7 | YTHDF1 YTHDF2 YTHDF3 YTHDC1 |
| ac4C |  | NAT10 | – | – |
m5C RNA methylation
m5C is another common RNA modification that involves the addition of a methyl group to the carbon-5 position of the cytosine base in RNA. m5c modifications can exist on mRNA, transfer RNA (tRNA), ribosomal RNA (rRNA), and long non-coding RNA (lncRNA), and are widely distributed across archaea, prokaryotes, and eukaryotes [20]. m5C participates in multiple stages of RNA metabolism, including splicing, stabilization, translation, maturation, and decay [21]. m5C expression relies on writers, such as NOP2/Sun domain (NSUN) RNA methyltransferase and DNA methyltransferase 2 (DNMT2), to catalyze m5C methylation [22, 23], while erasers, including tet methylcytosine dioxygenase 2 (TET2), demethylates m5C [24]. RNA binding protein ALY/REF export factor (ALYREF) recognizes m5C modifications on mRNA to promote mRNA nuclear output [22], while Y-box binding protein 1 (YBX1) binds directly to m5C modifications to stabilize mRNA [25]. In recent years, abnormal m5C modification and dysregulation of writers, erasers, and readers have played significant roles in the development and progression of various cancers, such as bladder, stomach, hepatocellular carcinoma (HCC), colorectal, prostate, breast, and lung cancer [23, 26]. However, further research is needed to identify the specific target genes and signal pathways that regulate m5C.
m7G RNA methylation
m7G is a highly conserved and prevalent post-transcriptional modification that plays a crucial role in regulating gene expression. In mRNA, m7G is primarily catalyzed by METTL1, a common methyltransferase, and forms a complex with WD repeat domain 4 (WDR4) [27]. In eukaryotes and a few archaea, m7G methylation at the mRNA 5ʹ cap is mediated by RNMT and RNMT-activating miniprotein (RAM) enzyme complex, that stabilize and protect the modified mRNA against exonuclization [28]. m7G influences various stages of mRNA processing and metabolism, including transcriptional stabilization, nuclear export, pre-mRNA splicing, translation, and polyadenylation [29, 30]. Additionally, m7G modifications have been shown to regulate the function of other RNA species, including rRNA and tRNA. The BUD23/TRMT112 methyltransferase complex catalyzes the formation of m7G at the G1639 site of 18s rRNA and induces 18s rRNA precursor biogenesis [31]. Dysregulation of m7G regulation can lead to aberrant m7G modification, promoting the development and progression of cancer.
m1A RNA methylation
In 1961, it was discovered that m1A modified the first nitrogen atom of RNA adenosine in mRNA, tRNA, rRNA, and lncRNA [32]. Although m1A modification is less common than m6A, both types of modifications share similar enzymes. It was found that nitrogen atoms could be transferred from m1A to m6A after Dimroth rearrangement in alkaline environments [33]. This suggests that RNA regulation may be dynamically alternating between m1A and m6A under certain conditions. m1A is controlled by methyltransferases (tRNA methyltransferase 6 (TRMT6), TRMT10C, TRMT61A, TRMT61B, and NML), demethylases (ALKBH1, ALKBH3, ALKBH7 and FTO), and readers (YTH N6-methyladenosine RNA binding protein 1–3 (YTHDF1-3) and YTHDC1). m1A regulates RNA stability and the initiation and extension of translation by modifying mRNA, tRNA, and rRNA [34]. Under physiological conditions, the methyl group in m1A carries a positive electrostatic charge which disrupts the base pairing with uracil [35]. Abnormal expression of m1A modifications may impact the biological function of RNAs and their potential interaction with partner proteins. Studies have found that aberrant m1A expression can contribute to a variety of diseases, including cardiovascular disease, lung disease, and Alzheimer's disease, as well as upregulate tumor cell proliferation, invasion, metabolism, and apoptosis [36–38].
ac4C RNA acetylation
N4-acetylcytidine (ac4C) is a highly conserved RNA acetylation modification found in eukaryotic and prokaryotic tRNAs, rRNAs, and mRNAs [39]. It plays a dominant role in the expression of genetic information and has been associated with a variety of conditions [40]. Previous studies have suggested that ac4C increases gene expression by regulating mRNA stability and translation efficiency, ensuring the accurate reading of codons and maintaining stability in tRNA [41]. Findings indicate that the location of the ac4c modification influences the function of RNA molecules. Insertion at helix 34 and 45 in rRNA was shown to promote self-biosynthesis and ribosome 40S subunit assembly and improve translation accuracy by enhancing mRNA stability [42, 43]. Conversely, ac4C expressed in the coding sequence (CDS) promoted translation extension, while insertion in the 5ʹ-untranslated region (5ʹ-UTR) inhibited the initiation of translation [44]. There is increasing evidence to suggest that ac4C plays an important role in the development, progression, and metastasis of cancer. The acetyltransferase NAT10, in conjunction with the adaptor protein THUMPD1 or attachment box C/D snoRNAs, is the only known protein that catalyzes the formation of ac4C [45]. Elevated NAT10 expression has been identified in various cancers across multiple organ systems, such as hepatocellular carcinoma, gastric, colon, pancreatic, cervical, bladder, esophageal, and lung cancer, with high levels significantly associated with increased malignant and aggressive characteristics of tumors [39].
RNA modifications in NSCLC
Lung cancer is the primary cause of cancer-related death worldwide [1, 46]. NSCLC is the dominant histopathological subtype of lung cancer [47], accounting for approximately 85% of lung cancers [48]. RNAs have been implicated in ineffective treatment strategies and the emergence of drug resistance, two major factors contributing to NSCLC tumor progression and poor prognosis. Therefore, in-depth research into the underlying mechanisms of NSCLC development and progression is critical to identify potential novel therapeutic targets.
m6A modification in NSCLC
m6A-dependent glycolysis in LUAD progression
Glycolysis is the primary pathway by which cells obtain energy in a low-oxygen environment. However, cancer cells exploit this process by reprogramming their metabolism to adapt to a hypoxic tumor microenvironment, a hallmark of malignancy [49, 50]. This metabolic shift increases glucose uptake, glycolysis, and lactic acid production, providing a continuous source of energy that fuels cancer cell proliferation, invasion, and migration [51]. Previous studies have linked m6A-dependent glycolysis to the progression of gastric, colorectal, and lung cancer [52, 53]. Ma et al. [54] recently reported significantly higher m6A levels in LUAD tumor tissue compared to adjacent healthy tissue. Further studies showed that m6A levels in LUAD tumor tissues were modulated by an upregulation of METTL3 and a downregulation of ALKBH5. LUAD mouse models demonstrated that the combination of increased METTL3 and decreased ALKBH5 had a strong tumor-promoting effect. Similarly, patients exhibiting simultaneous upregulation of METTL3 and downregulation of ALKBH5 had shorter survival times compared to patients expressing either molecule alone. The m6A modification reader YTHDF1 also plays a vital role in this process. High METTL3 and low ALKBH5 levels enhanced m6A methylation at position 359 A, facilitating the binding of ENO1 to YTHDF1 and enhancing ENO1 translation. ENO1 is a key enzyme involved in the catalyzation of 2-phosphoglycerate to produce phosphoenolpyruvate, promoting cytoplasmic glycolysis and promotion of LUAD progression (Fig. 1C). Therefore, targeting the m6A-dependant glycolytic pathway, particularly the ENO1-YTHDF1 complex, may be an effective therapeutic strategy for LUAD management.

Molecular mechanism of m6A modification involved in progression of NSCLC. A HNRNPA2B1 is increased in NSCLC and regulates lncRNA MEG3 through recognition of m6A, contributing to NSCLC tumorigenesis. B The m6A reader IGF2BP3 recognizes the m6A modification on MCM5 mRNA and increases MCM5 expression, thereby promoting the metastasis of LUAD cells. C Upregulation of the m6A writer METTL3, downregulation of m6A eraser ALKBH5, and upregulation of m6A reader YTHDF1 promote ENO1 mRNA translation by mediating m6A modification, thereby increasing cytoplasmic glycolysis and LUAD progression. D The IGF2BP3-COX6B2 axis mediates TKI resistance in NSCLC by promoting OXPHOS
TKI resistance in NSCLC
Metabolic reprogramming drives the growth, proliferation, and metastasis of cancer cells. This metabolic adaptation also represents a primary cause of treatment resistance in lung cancer [55]. Lin et al. [56] identified a novel mechanism underlying acquired resistance against epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). This mechanism was mediated by IGF2BP3, which regulated epigenetic modification and metabolic reprogramming by targeting downstream mRNA Cytochrome c oxidase subunit 6B polypeptide 2 (COX6B2). Research suggested that upregulation of IGF2BP3 expression reduced the sensitivity of TKI-resistant NSCLC to TKI therapy, consequently increasing drug resistance. This process of drug resistance is initiated by the binding of IGF2BP3 to the 3'-UTR of COX6B2 in an m6A-dependent manner, increasing the stability of COX6B2 and promoting oxidative phosphorylation (OXPHOS) (Fig. 1D). The IGF2BP3-COX6B2 axis also modulates niacinamide metabolism, thereby altering OXPHOS and driving EGFR-TKI-acquired drug resistance. In addition, inhibition of OXPHOS was found to enhance the efficacy of EGFR-TKIs, highlighting the role of OXPHOS in TKI-resistance. These results suggest that targeting IGF2BP3 may be an effective treatment strategy to improve the efficacy of EGFR-TKI therapies in NSCLC.
Angiogenesis in NSCLC
Angiogenesis is the formation of new blood vessels driven by vascular endothelial growth factor (VEGF) and is an essential component for tumor growth and metastasis [57]. Zhang et al. [58] recently revealed that m6A modification, mediated by high METTL3 expression or low ALKBH5 expression, accelerated lung cancer angiogenesis by promoting the translation of VEGF-A (Fig. 2B). Further research found that m6A-mediated VEGF-A translation was initiated by methylation of the internal ribosome entry site (IRES) at the VEGF-A mRNA 5ʹ-UTR site, leading to the recruitment of the YTHDC2/eIF4GI complex. In addition, TRPM7, regulated by METTL3 and IGF2BP2, has been associated with the proliferation, migration, and angiogenesis of NSCLC. Another molecular factor that is also regulated by METTL3 and IGF2BP2 and contributes to angiogenesis and metastasis is lncRNA deoxyguanosine kinase antisense RNA 1 (DGUOK-AS1). Feng et al. [59] showed that DGUOK-AS1 silencing reduced the stability of TRPM7 by modulating METTL3/IGF2BP2-mediated m6A modification, inhibiting proliferation, migration, and angiogenesis of NSCLC cells (Fig. 2A). This was the first time an association between DGUOK-AS1 and TRPM7 was found. Fang et al. [60] also reported that IGF2BP2 was highly expressed in LUAD tumor cells and was responsible for upregulating FLT4 expression by mediating m6A modification of FLT4 mRNA. Elevated FLT4 expression promoted angiogenesis and metastasis of LUAD by activating the PI3K–AKT pathway (Fig. 2C). Therefore, the IGF2BP2–FLT4–PI3K–AKT signaling pathway may play a key role in lung cancer progression and function as a prognostic marker, potentially highlighting an alternative research direction for NSCLC targeting drugs.

m6A regulatory protein affects lung cancer angiogenesis by regulating the expression of related proteins. A High expression of m6A writer METTL3 and reader IGF2BP2 influence lung cancer angiogenesis and metastasis by modulating TRPM7 expression. B High expression of METTL3 or low expression of ALKBH5 accelerates the angiogenesis of lung cancer by increasing the expression of VEGF-A. C The expression of IGF2BP2 increases in LUAD tumor cells and modulates the expression of FLT4 by mediating m6A modification. This promotes the angiogenesis and metastasis of lung adenocarcinoma by activating the PI3K–AKT pathway
m6A modification mediates ferroptosis in NSCLC
Ferroptosis is an iron-dependent mode of regulated cell death that inhibits cancer growth, characterized by an increase in internal reactive oxygen species (ROS) and lipid peroxidation [61]. Ferroptosis is regulated primarily by glutathione peroxidase 4 (GPX4), as well as m6A regulatory proteins, such as METTL3, IGF2BP3, FTO, and YTHDF2 [62–65]. Recent studies have shown that exosomal miR-4443 regulated the expression of anti-ferroptosis factor 1 (FSP1) in an m6A-dependent manner via METTL3 [62]. High expression of miR-4443 has been shown to negatively regulate METTL3, resulting in increased FSP1 levels, which in turn inhibited cisplatin-induced ferroptosis and promoted cisplatin resistance in NSCLC (Fig. 3B). In addition, Xu et al. [63] also found that high expression of IGF2BP3 in LUAD enhanced mRNA stability and expression level of various anti-ferroptosis factors, such as GPX4, SLC3A2, ACSL3, and FTH1, which inhibited ferroptosis and promoted LUAD progression (Fig. 3A). These findings highlight the role of m6A and associated regulatory proteins in modulating ferroptosis in NSCLC. Targeting proteins like METTL3 or IGF2BP3 may serve as a new therapeutic strategy for LUAD.

Mechanisms of ferroptosis mediated by m6A and m5C modifications in NSCLC. A High expression of IGF2BP3 in LUAD enhances the mRNA stability and expression level of anti-ferroptosis factor through m6A modification, inhibiting iron ferroptosis and promoting LUAD progression. B Exosome miR-4443 negatively regulates METTL3, promoting the expression of FSP1 and regulating iron death by regulating m6A modification. C m5C writer NSUN2 is highly expressed in NSCLC tumor cells, and m5C modification is mediated in the NRF2 mRNA 5ʹUTR region recognized by m5C reader YBX1. The stability of NRF2 mRNA is enhanced, leading to the upregulation of NRF2 expression and inhibiting ferroptosis
m6A RNA modification mediated by lncRNAs in NSCLC
Long non-coding RNAs (lncRNAs), previously thought to be non-functional genomic regions, have recently been found to play an important regulatory role in epigenetic modification, significantly influencing cellular processes [66]. LncRNAs are widely involved in the regulation of various cellular processes and can be affected by RNA methylation modification. Various RNA methylation, including m6A, m5C and m1A, can affect the folding, stability and interaction of lncRNA. Recent studies have shown that dysmethylation of lncRNA may lead to lung cancer and may be a potential biomarker or therapeutic target. Increased expression of IGF2BP2 was detected in post-operative NSCLC samples, and further studies identified lncRNA MALAT1 as its downstream target molecule. Mechanically, IGF2BP2 promotes the stability of lncRNA MALAT1 in an m6A-dependent manner, thereby promoting the expression of its downstream target ATG12 and proliferation of NSCLC [67]. LCAT1, a newly discovered lncRNA, promotes the growth of lung cancer cells both in vivo and in vitro. Further studies showed that LCAT1 prevented the autolysis of IGF2BP2 by interacting with IGF2BP2, thus making IGF2BP2 stable expression. The LCAT1/IGF2BP2 complex increases mRNA stability in an m6A-dependent manner, which in turn increases CDC6 levels [68]. CDC6 is a key cell cycle regulator. CDC6 levels were elevated in lung cancer tissues and correlated with patient survival. Therefore, the LCAT1-IGF2BP2-CDC6 axis may play an important regulatory role in the growth and migration of lung cancer cells and can be used as a potential therapeutic target for lung cancer. In addition, lncRNA LCAT3 expression was up-regulated in LUAD and correlated with poor prognosis in patients. METTL3-mediated m6A modification increased the stability of LCAT3 and up-regulated its expression. Highly expressed LCAT3 activates MYC transcription through recruitment of upstream element binding protein 1(FUBP1), thereby promoting the proliferation, invasion and metastasis of lung cancer cells [69]. LCAT3 has shown strong carcinogenic activity, so targeting the LCAT3-FUBP1-C-MYC axis may be a promising therapeutic strategy for LUAD.
Other m6A modifications in NSCLC
m6A modification is one of the most common mammalian methylation modifications and is involved in the progression of many cancers, including NSCLC. HNRNPA2B1, an m6A reader, has been shown to display increased expression in NSCLC tumor tissues compared to healthy tissues, and inhibition of HNRNPA2B1 decreased the m6A level in lncRNA MEG3. lncRNA MEG3 has exhibited protective anti-cancer functions, as its knockdown resulted in the promotion of cell proliferation and invasion (Fig. 1A). Therefore, HNRNPA2B1 may be an NSCLC-specific oncogene through m6A-dependent modification of lncRNA MEG3 [70].
Another m6A reader is IGF2BP3, a member of the IGF2BP family. Yang et al. [71] found that IGF2BP3 was highly expressed in LUAD tumor tissues, where it recognized mRNA m6A modifications of the minichromosome maintenance protein 5 (MCM5) and enhanced mRNA stability. MCM5, an evolutionarily conserved member of the MCM family, plays a key role in loading DNA onto the replication starting point [72]. Increased MCM5 levels have been shown to competitively inhibit SIRT1-mediated deacetylation of the Notch1 intracellular domain (NICD1), resulting in NICD1 protein stabilization. This activation of the IGF2BP3/MCM5/NICD1 axis promoted m6A-dependent IGF2BP3-mediated tumor cell plasticity, facilitating LUAD tumor cell metastasis (Fig. 1B). METTL3, a writer of m6A, has been found to promote the angiogenesis of lung cancer cells by targeting VEGF-A [58] and has also been implicated in LUAD tumorigenesis by specifically targeting ENO1 to regulate glycolysis within the tumor microenvironment [54].
m5C modification in NSCLC
Development of EGFR-TKIs drug resistance in patients with EGFR mutant NSCLC
EGFR-targeted therapy is the latest first-line treatment for patients with advanced NSCLC who have developed EGFR-activated mutations [73]. However, due to the inherent resistance to EGFR-TKIs, approximately 30% of patients have poor treatment response and poor recovery [74]. The development of the EGFR-mutant genotype is a significant challenge in the treatment of NSCLC, leading to poor prognosis and reduced treatment efficacy [75, 76]. Currently, known risk factors for EGFR-TKIs acquired resistance are secondary T790M mutation, MET amplification, HER2 amplification, and transformation into SCLC. However, little has been reported regarding the role and mechanism of RNA m5C modifications. Wang et al. [77] has suggested that RNA m5C methylation modification in NSCLC tumor drug resistance was mediated by the NSUN2-YBX1-QSOX1 axis (Fig. 4C). NSUN2 expression levels were elevated in NSCLC, and overexpression of NSUN2 modulated QSOX1 protein levels in an MFC-YBX1-dependent manner. The expression of QSOX1 was correlated with endogenous gefitinib resistance. Therefore, the NSUN2-YBX1-QSOX1 axis may act as a potential candidate biomarker for prognosis and therapeutic target in NSCLC patients with intrinsic resistance to EGFR-TKIs.

Molecular mechanisms of m5C modification involved in the progression of NSCLC. A YBX1 recognizes m5C methylation in PFKFB4 mRNA and induces an increase in PFKFB4 expression, thus promoting the progression of LUSC. B NSUN2 enhances the stability of downstream target PIK3R2 mRNA by regulating m5C modification, thereby activating the PI3K–AKT signal and promoting the malignant progression of LUAD. C The NSUN2-YBX1-QSOX1 axis mediates TKI resistance in NSCLC patients. D LINC02159 binds to ALYREF and activates downstream Hippo and Wnt/β-catenin pathways by mediating m5C to upregulate YAP1 expression, contributing to carcinogenesis
m5C RNA modification mediated by lncRNAs in NSCLC
lncRNAs, which control gene expression through various mechanisms, are considered key regulators in human health and disease [78]. There is increasing evidence to suggest that lncRNAs regulate gene expression and post-transcriptional regulation. Yang et al. [79] discovered a novel lncRNA, LINC02159, that was significantly upregulated in NSCLC tumor tissues compared to healthy tissues. Further evaluation showed that LINC02159 promoted the development of non-small cell tumors by binding to ALYREF which enhanced m5C modification of YAP1 mRNA, and subsequently increased YAP1 mRNA expression (Fig. 4D). YAP1 upregulation activated downstream Hippo and β-catenin pathways which have been closely associated with cancer cell growth and metastasis. Therefore, the LINC02159-ALYREF-YAP1 axis may serve as a potential biomarker and therapeutic target for NSCLC patients.
Ferroptosis resistance in NSCLC
Ferroptosis is a mechanism that can be utilized as a novel anticancer treatment. Excess-free iron (Fe2+) stimulates the production of ROS through a Fenton reaction, inducing cell death [80]. NF-E2-related factor 2 (NRF2) is a core component that inhibits ferroptosis under oxidative stress conditions by enhancing the transcription of antioxidants, such as FTH1 and GPX4 [81, 82]. However, Chen et al. [83] found that NSCLC patients exhibited dysregulated NRF2 expression levels, with higher expression correlated with poorer prognosis. As previously mentioned, NSUN2 is also highly expressed in NSCLC tumor cells, and m5C modification in the NRF2 mRNA 5ʹUTR region, recognized by YBX1, can lead to increased stability and upregulation of NRF2 mRNA, subsequently inhibiting ferroptosis (Fig. 3C). Therefore, targeting NSUN2 and the ferroptosis pathway may provide a promising therapeutic strategy for NSCLC patients.
Other m5C modifications in NSCLC
mRNA nuclear export is a key step in gene expression as it enables the transport of mature mRNAs, mediated by the transcription–export (TREX) complex, from the nucleus to the cytoplasm where they can be translated into proteins. This is a tightly regulated process that can significantly impact the growth of eukaryotic cells by regulating the expression of key genes responsible for proliferation, differentiation, and survival. The THO complex (THOC), a component of the TREX complex, is comprised of six subunits: THOC1-3 and THOC5-7 [84], and influences the splicing, extension, and nuclear export of nascent RNA. Recent studies have shown that THOC3 is highly expressed in LUSC, with implications for cancer cell growth, migration, and glycolysis. Several other molecules are involved in driving LUSC carcinogenesis, such as PFKFB4. PFKFB4 is a protein associated with cancer cell survival by regulating glycolysis and pentose phosphate production, and its inhibition leads to cell cycle arrest and glycolytic inhibition [85]. Further studies sought to elucidate the interaction between PFKFB4 and THOC3. Researchers found that when PFKFB4 mRNA exited the nucleus, THOC3 recruited YBX1 which recognizes m5C methylation of PFKFB4 and synergically induced PFKFB4 expression, promoting the progression of LUSC [86] (Fig. 4A). Potential therapeutic targets include the RNA methyltransferase, NSUN2, which has potential roles in cellular proliferation and senescence. Du et al. [87] found that NSUN2 enhanced the stability of phosphoinositide-3-kinase regulatory subunit 2 (PIK3R2) mRNA by promoting m5C methylation, thereby advancing malignant characteristics of lung adenocarcinoma via PI3K–AKT signaling pathway activation (Fig. 4B). These findings suggest that targeting THOC3 and NSUN2 may be novel biomarkers and promising therapeutic targets for patients with LUAD.
m7G modification in lung cancer
It has been reported that m7G regulatory proteins are upregulated and promote tumor progression in several cancers, including prostate cancer, bladder cancer, liver cancer, and lung cancer. However, the association between m7G regulatory proteins and tumor progression in LUAD patients has not been fully elucidated. Research has shown that a methyltransferase complex consisting of WDR4 and METTL1 catalyzes eukaryotic tRNA m7G modification, promoting the translation of cell cycle-related mRNAs and the growth and invasion of lung cancer [88]. In vitro analysis corroborated the association between the complex and cancer development, finding that knockdown of the WDR4-METTL1 complex slowed the progression of lung cancer cells. In addition, the m7G-related gene, NUDT4, promoted the proliferation of cancer cells in lung adenocarcinoma. While the specific mechanism underlying the anti-proliferative effects is not fully understood, NUDT4 may act as a novel target to inhibit the proliferation of tumor cells [89]. In addition, an m7G-Riskscore of LUAD was generated that incorporated m7G genetic alterations in the tumor microenvironment, such as CAND1, RRM2, and SLC2A1, to predict the overall survival (OS) of patients [90]. Analysis revealed that patients exhibiting a higher m7G-Riskscore had shorter OS, and m7G-Riskscore was highly correlated with clinical characteristics and prognosis of tumors, potentially highlighting its utility as an independent prognostic factor. Other studies have found that METTL1 promoted the formation of m7G methylation on VEGF-A mRNA, leading to an increase in VEGF-A translation, which in turn promoted tumor angiogenesis [91]. These findings highlight the role of m7G and associated regulatory proteins in the formation and progression of cancer and possible targets for therapy. However, the mechanisms underlying LUAD progression require further investigation.
m1A modification and lung cancer
ALKBH3, a member of the AlkB family and a demethylase targeting m1A, has been found to be involved in lung cancer tumorigenesis. Tasaki et al. [92] found that ALKBH3 is highly expressed in lung adenocarcinoma cells, with a significant negative correlation between expression profile and relapse-free survival (RFS). Further studies showed that ALKBH3 promoted the growth of lung adenocarcinoma cells by accelerating the G1/S transition and driving cellular proliferation. ALKBH3 gene silencing using siRNA transfection effectively elicited cellular senescence and inhibition of lung adenocarcinoma cell growth by inducing expression of p21/p27, highlighting the specific role of ALKBH3 in lung cancer and its therapeutic potential [92]. In addition, Zhou et al. [93] developed a comprehensive scoring system, termed Writer-Score, that integrated four different types of RNA modifications to evaluate the effect of neoadjuvant immunotherapy in patients with NSCLC. Subsequent research discovered a strong correlation between Writer-Score and the expression level of PD-L1. Primarily used to predict the prognosis of NSCLC patients who receive neoadjuvant PD-L1 inhibitors, the Writer-Score demonstrated high prognostic efficacy in NSCLC patients receiving neoadjuvant immunotherapy [93]. These findings highlight the potential utility of targeting m1A-associated regulatory proteins to mitigate the progression of lung cancer.
ac4C modification and lung cancer
NAT10 is the only known RNA acetylase that catalyzes the formation of ac4C modification on RNA. Studies have found that NAT10 expression was upregulated in various cancers, such as colon cancer, liver cancer, and lung cancer [39, 94, 95]. Recent evidence has suggested that ac4C mRNA expression was significantly higher in LUAD tumor tissues compared to healthy adjacent tissues. Notably, ac4c modifications were predominantly concentrated within the coding sequence, suggesting that ac4c may play a role in a wide range of cellular functions in LUAD [96]. In addition, researchers previously identified an increase in ac4c expression of transcription factor AP-2 alpha (TFAP2A) mRNA in LUAD [97]. Similarly, Shang et al. [96] associated TFAP2A with the progression of cancer through the activation of pro-metastatic pathways. Therefore, increased expression of TFAP2A may serve as a reliable predictor of poor prognosis in lung cancer patients. However, the precise mechanisms of TFAP2A remain to be elucidated.
MicroRNAs (miRNAs) are single-stranded RNA sequences approximately 21 nucleotides in length that play key roles in almost every biological process. Biogenesis of miRNA relies on primary miRNAs (pri-miRNA) and a range of protein interactions. Modification to an miRNA sequence, such as ac4C, can influence subsequent processing, stability, and gene silencing efficiency. NAT10 and THUMPD1 are molecules that influence miRNA production by regulating pri-miRNA ac4C expression, which plays a crucial role in the occurrence and development of tumors [98]. Overexpression of NAT10 has been shown to increase the level of ac4C of pri-miRNA, thereby enhancing interaction with DGCR8, a protein involved in miRNA biogenesis, promoting the processing of pri-miRNA into precursor miRNA (pre-miRNA) by microprocessor DROSHA/DGCR8. Accumulation of pre-miRNA subsequently increases the level of mature miRNA, contributing to the development of cancer. In addition, Ruhul Amin et al. [99] found that NAT10 promoted metastasis of breast tumor cells to the lung. Further research highlighted that overexpression of NAT10 in tumor cells promoted the transcription of metastasis-related genes, including chemokines and cytokines, in a P300-dependent manner. This production of pro-metastatic molecules promoted the recruitment of neutrophils and monocytes, exacerbating tumor cell metastasis. The loss of NAT10 was found to trigger the misalignment of p300 and the destruction of associated enhancers, suppressing the transcription of genes required for tumor cell metastasis. This highlights the potential role of ac4C modification and NAT10 as novel molecular markers and therapeutic targets for lung cancer. However, the molecular mechanism of ac4C modification involved in lung cancer progression remains unclear, and further studies are needed.
RNA modification and SCLC
SCLC is a highly malignant neuroendocrine tumor that accounts for approximately 10–15% of lung cancers [100]. SCLC has a severely poor prognosis, with a median OS for standard chemotherapy regimens less than 1 year due to the high incidence of primary or secondary radiochemotherapy insensitivity [101, 102]. Therefore, exploration into the molecular mechanisms that contribute to tumor drug resistance can provide novel therapeutic options for SCLC and improve the clinical prognosis.
Epigenetics has been implicated in the progression of SCLC, playing a vital role in tumor metastasis and insensitivity to chemoradiotherapy [103, 104]. Epigenetic alterations located in the promoter region of human telomerase reverse transcriptase (hTERT) have been widely reported as common non-coding genomic modifications in a variety of cancers [105, 106], including lung cancer. Upregulation of hTERT can promote the growth, proliferation, and migration of cancer cells. In addition, high levels of radiation-induced methylation of the hTERT promoter region can lead to radiotherapy resistance in SCLC through upregulation of its downstream target EZH2. To clarify the role of the m6A regulatory factor in SCLC, Zhang et al. [107] constructed a stratified prognostic risk score (m6A score). The m6A score was shown to accurately predict OS, RFS, chemotherapy efficacy, and immunotherapy response of SCLC patients, highlighting its potential utility as a prognostic tool. However, the role of m6A regulators in SCLC is still not fully known, and further studies are needed. Recent studies have found that m6A writer METTL3 was highly expressed in SCLC, and its expression was associated with chemotherapy resistance in SCLC [108]. Further studies also found that METTL3 promoted chemotherapy resistance in SCLC by inducing mitochondrial autophagy, a type of selective autophagy required for controlling mitochondrial quality. Under stimulation conditions, mitochondria participate in various biological processes by sensing a variety of factors, including extracellular signals and depolarization of membrane potential [109, 110]. Other research has demonstrated that METTL3 promoted mitochondrial autophagy by inducing m6A methylation on DCP2 mRNA, an important 5ʹ-decapping enzyme. This resulted in decreased expression of DCP2 protein, activating the downstream Pink1–Parkin pathway that initiated mitochondrial autophagy and subsequently induced chemotherapy resistance in SCLC (Fig. 5). In addition, other studies found that SCLC patients with low expression of m6A reading protein YTHDF2 have longer survival times, potentially related to YTHDF2 negative regulation of immune infiltration [111]. This suggests that immunotherapy may be suitable for SCLC patients with low YTHDF2 expression. Therefore, m6A-associated molecules, such as YTHDF2, METTL3, and DCP2, may serve as potential biomarkers for SCLC tumor detection, treatment, prognosis, and chemotherapy resistance.

METTL3 induces m6A methylation of DCP2, degrading DCP2 and promoting mitochondrial autophagy through the Pink1–Parkin pathway, leading to chemotherapy resistance in SCLC
Conclusion and future prospects
Epigenetic modifications, specifically RNA modifications such as m6A, m5C, m7G, m1A, and ac4C in mRNA, tRNA, and rRNA, have been widely implicated as major factors contributing to lung cancer. These modifications are catalyzed by regulatory proteins, especially “writer” molecules, which modulate the expression of downstream genes and influence the biological function of tumor cells (summarized in Table 2). Various RNA modifications have been shown to impact tumor progression and drug resistance by regulating key cellular processes, such as glycolysis, ferroptosis, and mitochondrial autophagy. Regulatory proteins regulate RNA modification, affect the stability or expression of mRNA, enhance the expression of cancer-promoting transcripts, and reduce the expression of cancer-inhibiting transcripts, thus promoting the occurrence and progression of lung cancer. Therefore, regulatory proteins have great potential to be used as new biomarkers and therapeutic targets for the diagnosis and treatment of lung cancer, for the early diagnosis and targeted therapy of lung cancer, to improve the prognosis of lung cancer patients and improve the survival rate. For patients with drug-resistant lung cancer, the study showed an even more significant effect. The research on regulatory proteins and their target molecules is expected to provide new targeted therapeutic drugs for drug-resistant patients. This review summarized the current understanding of molecular mechanisms of RNA modifications in lung cancer development, progression, metastasis, and resistance to targeted therapies, highlighting their potential as novel therapeutic targets. While m6A modification has been extensively studied, research on other modifications remains in the preliminary stage due to limitations in detection methods and the complexity of underlying mechanisms. Further research is required to fully elucidate the role of RNA modifications in lung cancer and explore the potential clinical implications.
Table 2
Functions of modified oncogenes in lung cancers
| Tumor | Modification | Regulatory protein | Target | Functions | References |
|---|---|---|---|---|---|
| NSCLC | m6A | HNRNPA2B1 | lncRNA MEG3 | Facilitate NSCLC tumorigenesis and metastasis | [70] |
| IGF2BP3 | MCM5 | Promote LUAD metastasis | [71] | ||
| METTL3 | VEGFA | Promote lung cancer cell angiogenesis | [58] | ||
METTL3 ALKBH5 YTHDF1 | ENO1 | Promote LUAD glycolysis and tumorigenesis | [54] | ||
| IGF2BP3 | COX6B2 | Promote acquired EGFR-TKI resistance | [56] | ||
| METTL3 IGF2BP2 | TRPM7 | Involved in proliferation, migration, and angiogenesis of NSCLC cells | [59] | ||
| IGF2BP2 | FLT4 | Promote LUAD angiogenesis and metastasis | [60] | ||
| IGF2BP2 | lncRNA MALAT1 | Promote NSCLC proliferation | [67] | ||
| IGF2BP2 | lncRNA LCAT1 | Promote tumor growth and migration | [68] | ||
| METTL3 | lncRNA LCAT3 | Promote the proliferation, invasion and metastasis of LUAD | [69] | ||
| IGF2BP3 | GPX4 SLC3A2 ACSL3 FTH1 | Inhibit ferroptosis of LUAD tumor cells | [63] | ||
| m5C | YBX1 | PFKFB4 | Facilitate proliferation, migration, and glycolysis of LUSC | [86] | |
| NSUN2 | PIK3R2 | Promote tumor growth and aggressiveness | [87] | ||
| NSUN2/YBX1 | QSOX1 | Involved in gefitinib resistance and tumor recurrence | [77] | ||
| ALYREF | YAP1 | Promote NSCLC progression | [79] | ||
| NSUN2/YBX1 | NRF2 | Promote lung cancer ferroptosis tolerance, proliferation, and migration | [83] | ||
| m7G | METTL1 WDR4 | tRNA | Promote lung cancer growth and invasion | [88] | |
| METTL1 | VEGFA | Promote lung cancer angiogenesis | [91] | ||
| – | NUDT4 | Promote lung cancer cell proliferation | [89] | ||
| m1A | ALKBH3 | – | Promote LUAD cell growth | [92] | |
| ac4C | – | TFAP2A | Promote lung cancer cell proliferation | [97] | |
NAT10 THUMPD1 | pri-miRNA | Modulate tumor progression | [98] | ||
| NAT10 | – | Promote lung metastasis of breast tumor cells | [99] | ||
| SCLC | m6A | METTL3 | DCP2 | Induce mitochondrial autophagy and promotes chemotherapy resistance | [108] |
Modification the type of RNA modification in lung cancer; Regulatory protein protein which can regulate RNA modification, actually is writer, reader or eraser of RNA modification; Target targets of regulatory protein; Functions the role of regulatory protein and its target in lung cancer; NSCLC non-small cell lung cancer; SCLC small cell lung cancer; m6A N6-methyladenosine; m5C 5-methylcytidine; m7G N7-methylguanosine; m1A N1-methyladenosine; ac4C N4-acetylcytidine
Abbreviations
| SCLC | Small cell lung cancer |
| NSCLC | Non-small cell lung cancer |
| LUSC | Lung squamous cell carcinoma |
| LUAD | Lung adenocarcinoma |
| LCC | Large cell carcinoma |
| m6A | N6-Methyladenosine |
| m5C | 5-Methylcytidine |
| m7G | N7-Methylguanosine |
| m1A | N1-Methyladenosine |
| mRNA | Messenger RNA |
| WER | Writing-erasing-reading |
| METTL3 | Methyltransferase-like protein 3 |
| RBM15 | RNA-binding motif protein 15 |
| ZC3H13 | Zinc finger CCCH-type containing 13 |
| WTAP | Wilms Tumor 1 associated protein |
| ALKBH5 | AlkB homolog 5 |
| FTO | FTO-alpha-ketoglutarate-dependent dioxygenase |
| IGF2BP | Insulin-like growth factor 2 mRNA binding proteins |
| hnRNP | Heterogeneous nuclear ribonucleoprotein |
| RRMs | RNA recognition motifs |
| tRNA | Transfer RNA |
| rRNA | Ribosomal RNA |
| lncRNA | Long non-coding RNA |
| NSUN | NOP2/Sun domain |
| DNMT2 | DNA methyltransferase 2 |
| TET2 | Tet methylcytosine dioxygenase 2 |
| ALYREF | ALY/REF export factor |
| YBX1 | Y-box binding protein 1 |
| HCC | Hepatocellular carcinoma |
| WDR4 | WD repeat domain 4 |
| RAM | RNMT-activating miniprotein |
| TRMT6 | TRNA methyltransferase 6 |
| ac4C | N4-Acetylcytidine |
| CDS | Coding sequence |
| 5ʹ-UTR | 5ʹ-Untranslated region |
| MCM5 | Minichromosome maintenance protein 5 |
| NICD1 | Notch1 intracellular domain |
| EGFR | Epidermal growth factor receptor |
| TKIs | Tyrosine kinase inhibitors |
| COX6B2 | Cytochrome c oxidase subunit 6B polypeptide 2 |
| OXPHOS | Oxidative phosphorylation |
| VEGF | Vascular endothelial growth factor |
| IRES | Internal ribosome entry site |
| DGUOK-S1 | Deoxyguanosine kinase antisense RNA 1 |
| ROS | Reactive oxygen species |
| GPX4 | Glutathione peroxidase 4 |
| FSP1 | Anti-ferroptosis factor 1 |
| TREX | Transcription–export |
| THOC | THO complex |
| PIK3R2 | Phosphoinositide-3-kinase regulatory subunit 2 |
| NRF2 | NF-E2-related factor 2 |
| OS | Overall survival |
| RFS | Relapse-free survival |
| TFAP2A | Transcription factor AP-2 alpha |
| miRNAs | MicroRNAs |
| pri-miRNA | Primary miRNAs |
| pre-miRNA | Precursor miRNA |
| hTERT | Human telomerase reverse transcriptase |
| FUBP1 | Far-Upstream Element Binding Protein 1 |
Author contributions
X.G. conceived of the study and designed the headings. S.Z., Y.L. and K.L. wrote and revised the manuscript text. S.Z. created the figure and table. X.H. supervised the study. All authors read and approved the final manuscript.
Funding
This study was sponsored by the National Natural Science Foundation of China (No 81600512) and Science and technology Research program of Henan Province (No 242102311156).
Availability of data and materials
No datasets were generated or analysed during the current study.
Declarations
Not applicable.
All authors have read the manuscript and have given their consent for publication.
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shujun Zhang and Yafeng Liu contributed equally to this work.
References
Articles from Cancer Cell International are provided here courtesy of BMC
Citations & impact
This article has not been cited yet.
Impact metrics
Alternative metrics

Discover the attention surrounding your research
https://www.altmetric.com/details/169723820
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
A novel m6A/m5C/m1A/m7G-related classification and risk signature predicts prognosis and reveals immunotherapy inclination in gastric cancer.
Transl Cancer Res, 13(7):3285-3298, 26 Jul 2024
Cited by: 0 articles | PMID: 39145046 | PMCID: PMC11319980
Involvement of RNA methylation modification patterns mediated by m7G, m6A, m5C and m1A regulators in immune microenvironment regulation of Sjögren's syndrome.
Cell Signal, 106:110650, 17 Mar 2023
Cited by: 6 articles | PMID: 36935085
Characterization of the m6A/m1A/m5C/m7G-related regulators on the prognosis and immune microenvironment of glioma by integrated analysis of scRNA-seq and bulk RNA-seq data.
J Gene Med, 26(2):e3666, 01 Feb 2024
Cited by: 1 article | PMID: 38391150
The role of RNA modification in hepatocellular carcinoma.
Front Pharmacol, 13:984453, 02 Sep 2022
Cited by: 12 articles | PMID: 36120301 | PMCID: PMC9479111
Review
 Free full text in Europe PMC
Free full text in Europe PMC
Funding
Funders who supported this work.
National Natural Science Foundation of China (1)
Grant ID: 81600512
Science and Technology Innovation Talents in Universities of Henan Province (1)
Grant ID: 242102311156


