Abstract
Free full text
An alternative UPF1 isoform drives conditional remodeling of nonsense‐mediated mRNA decay
 1
1
Abstract
The nonsense‐mediated mRNA decay (NMD) pathway monitors translation termination in order to degrade transcripts with premature stop codons and regulate thousands of human genes. Here, we show that an alternative mammalian‐specific isoform of the core NMD factor UPF1, termed UPF1LL, enables condition‐dependent remodeling of NMD specificity. Previous studies indicate that the extension of a conserved regulatory loop in the UPF1LL helicase core confers a decreased propensity to dissociate from RNA upon ATP hydrolysis relative to UPF1SL, the major UPF1 isoform. Using biochemical and transcriptome‐wide approaches, we find that UPF1LL can circumvent the protective RNA binding proteins PTBP1 and hnRNP L to preferentially bind and down‐regulate transcripts with long 3’UTRs normally shielded from NMD. Unexpectedly, UPF1LL supports induction of NMD on new populations of substrate mRNAs in response to activation of the integrated stress response and impaired translation efficiency. Thus, while canonical NMD is abolished by moderate translational repression, UPF1LL activity is enhanced, offering the possibility to rapidly rewire NMD specificity in response to cellular stress.
core NMD factor UPF1, termed UPF1LL, enables condition‐dependent remodeling of NMD specificity. Previous studies indicate that the extension of a conserved regulatory loop in the UPF1LL helicase core confers a decreased propensity to dissociate from RNA upon ATP hydrolysis relative to UPF1SL, the major UPF1 isoform. Using biochemical and transcriptome‐wide approaches, we find that UPF1LL can circumvent the protective RNA binding proteins PTBP1 and hnRNP L to preferentially bind and down‐regulate transcripts with long 3’UTRs normally shielded from NMD. Unexpectedly, UPF1LL supports induction of NMD on new populations of substrate mRNAs in response to activation of the integrated stress response and impaired translation efficiency. Thus, while canonical NMD is abolished by moderate translational repression, UPF1LL activity is enhanced, offering the possibility to rapidly rewire NMD specificity in response to cellular stress.
Abstract
UPF1LL targets a subset of mRNAs for degradation by overcoming the substrate‐shielding binding of PTBP1 and hnRNP L.

Introduction
Nonsense‐mediated mRNA decay (NMD) is an evolutionarily conserved mRNA quality‐control pathway that degrades transcripts undergoing premature translation termination (Smith & Baker, 2015; Lavysh & Neu‐Yilik, 2020). In addition, NMD performs a regulatory role by governing the turnover of ~5–10% of the transcriptome, including mRNAs with upstream open reading frames (uORFs), introns downstream of the stop codon, or long 3’ untranslated regions (UTRs) (Kishor et al, 2019a). Despite extensive studies of the large impact of NMD on the transcriptome, the mechanisms by which the pathway selects its regulatory targets are poorly understood.
A suite of conserved NMD factors acts in concert with general mRNA binding proteins and RNA decay enzymes to identify and degrade target mRNAs. The RNA helicase UPF1 is a central coordinator of the NMD pathway, as it directly binds mRNA and functions at multiple steps in the selection and degradation of target transcripts (Kim & Maquat, 2019). Additional core NMD factors UPF2 and UPF3 promote UPF1 activity and link UPF1 to the exon junction complex (EJC), which strongly stimulates decay (Le Hir et al, 2000a, 2000b; Chamieh et al, 2008). In many eukaryotes, NMD execution also depends on the SMG1, 5, 6, and 7 proteins (Hodgkin et al, 1989; Pulak & Anderson, 1993; Page et al, 1999; Causier et al, 2017). Phosphorylation of UPF1 by the SMG1 kinase is required for efficient mRNA decay (Kashima et al, 2006), as phosphorylated UPF1 recruits the SMG6 endonuclease and/or general decapping and deadenylation enzymes through the SMG5/7 heterodimer (Huntzinger et al, 2008; Eberle et al, 2009; Loh et al, 2013).
In addition to the functions of specialized NMD proteins, substrate selection and degradation by the NMD pathway require the translation termination machinery to detect in‐frame stop codons (Karousis & Mühlemann, 2019). Although the molecular details remain to be elucidated, it is widely accepted that interactions between core NMD factors and a terminating ribosome are necessary for decay (Lavysh & Neu‐Yilik, 2020). Because of the strict dependence of NMD on translation termination, decay efficiency of canonical NMD targets is expected to be tightly linked to translation efficiency. However, there is evidence that NMD efficiency for some targets is actually enhanced when translation is impaired by treatment with the mTOR inhibitor rapamycin or the translation elongation inhibitor emetine (Martinez‐Nunez et al, 2017). These data warrant a more extensive investigation into the role of translation in shaping target specificity by the NMD pathway, particularly during changing physiological conditions.
The ability of UPF1 to bind and hydrolyze ATP is critical for the selection and degradation of potential NMD substrates (Franks et al, 2010; Kurosaki et al, 2014; Lee et al, 2015). Numerous studies have provided evidence that the affinity of UPF1 for RNA is reduced by ATP binding and hydrolysis, in a manner dependent on an 11 amino acid regulatory loop in domain 1B of the helicase core that protrudes into the RNA binding channel (Czaplinski et al, 1995; Weng et al, 1998; Cheng et al, 2007; Chamieh et al, 2008; Chakrabarti et al, 2011; Fiorini et al, 2013; Gowravaram et al, 2018). Intriguingly, mammals undergo an alternative splicing event to express two UPF1 isoforms that differ only in length of the regulatory loop (Fig1A). Almost all NMD studies to date have focused on the more abundant UPF1 “short loop” isoform (designated herein UPF1SL), which contains the 11 amino acid regulatory loop that most potently weakens the affinity of UPF1 for RNA in the presence of ATP. Alternative 5’ splice site usage in exon 7 of UPF1 generates a second UPF1 isoform that extends the regulatory loop to 22 amino acids, the sequence of which is conserved among mammals spanning humans to marsupials (Appendix Fig S1A). This naturally occurring UPF1 “long loop” isoform (designated herein UPF1LL), which represents ~15–25% of total UPF1 mRNA in diverse cell and tissue types (Appendix Fig S1B), has increased catalytic activity and a higher affinity for RNA in the presence of ATP than the UPF1SL isoform (Gowravaram et al, 2018). It is unknown whether the differential biochemical properties of the UPF1LL isoform affect NMD specificity in cells.

(Top) Schematic representation of alternative 5' splice site usage in exon 7 of mammalian UPF1 that results in two UPF1 protein isoforms that differ in length of the regulatory loop within the helicase core. (Bottom) Ribbon diagram of human UPF1SL helicase core overlaid with that of human UPF1LL. The regulatory loop in domain 1B is indicated for UPF1SL (light blue) and UPF1LL (purple), based on Protein Data Bank accessions 2XZP and 6EJ5 (Chakrabarti et al, 2011; Gowravaram et al, 2018).
(Top) Semiquantitative RT–PCR of UPF1SL or UPF1LL transcript levels following transfection of HEK‐293 cells with a NT siRNA or a siRNA that specifically targets the UPF1LL isoform. (Bottom) Venn diagram (to scale) of overlapping targets identified from RNA‐seq following total UPF1 or UPF1LL‐specific knockdown. Depicted are genes that increased in abundance at least 1.4‐fold (FDR
<0.05) and met read count cutoffs in both datasets. P‐value indicates enrichment of genes that increased in abundance at least 1.4‐fold (FDR<0.05) with UPF1LL‐specific knockdown among those regulated by total UPF1, as determined by Fisher's exact test.Heat map of changes in relative mRNA abundance for genes encoding NMD factors, as determined from RNA‐seq following transfection of HEK‐293 cells with a siRNA that targets both UPF1 isoforms (UPF1total) or a siRNA that specifically targets the UPF1LL isoform.
Density plot of changes in relative mRNA abundance as determined by RNA‐seq in SMG7ko/SMG6kd cells, relative to a parental cell line treated with control siRNAs (Boehm et al, 2021). Genes were categorized as up‐regulated by siUPF1total only, siUPF1LL only, or both siUPF1total and siUPF1LL. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
RT–qPCR analysis of indicated transcripts following transfection of HEK‐293 cells with the indicated siRNAs. Relative fold changes are in reference to NT siRNA. Asterisk (*) indicates P
<0.05, as determined by two‐way ANOVA. Black dots represent individual data points and error bars indicate mean±SD (n=3 biological replicates). Dashed line indicates log2(fold change) of +0.5. PTC+ indicates the use of primers specific to transcript isoforms with validated poison exons (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P‐values associated with each statistical comparison.Gene ontology analysis of 1621 genes that increased in expression at least 1.4‐fold upon UPF1LL‐specific knockdown in HEK‐293 cells under normal cellular conditions. Genes may map to multiple categories.
Source data are available online for this figure.
Here, we show that the UPF1LL isoform gives the mammalian NMD pathway the latent ability to remodel NMD target specificity in response to changing physiological conditions. We identify that UPF1LL can overcome inhibition by polypyrimidine tract binding protein 1 (PTBP1) and heterogeneous nuclear ribonucleoprotein L (hnRNP L) to preferentially associate with and down‐regulate long 3’UTRs normally shielded from NMD. Unexpectedly, we find that UPF1LL activity is sustained or even enhanced in conditions of reduced translation efficiency, including during the integrated stress response. mRNAs subject to UPF1LL‐dependent downregulation upon translation inhibition include hundreds of mRNAs not normally targeted by NMD, many of which are protected by PTBP1 and hnRNP L. Together, our data support that human cells use the UPF1LL isoform to conditionally alter which mRNAs are selected and degraded by the NMD pathway, expanding the scope of NMD in mammalian gene expression control.
Results
Specific UPF1LL depletion causes alterations in gene expression
To specifically interrogate the cellular functions of the UPF1LL isoform, we developed a siRNA that efficiently degrades UPF1LL mRNA without perturbing the expression of the major UPF1SL isoform (Fig1B (top) and FigEV1A). As an initial analysis of UPF1LL functions, we treated human HEK‐293 cells with the UPF1LL‐specific siRNA (siUPF1LL) and performed total RNA‐seq. Differential expression analysis identified 1621 genes that were at least 1.4‐fold more highly expressed upon UPF1LL knockdown, out of a total population of 13,668 genes analyzed, indicating a role for endogenous UPF1LL in gene expression regulation (Dataset EV1). To investigate whether the observed changes in mRNA abundance with UPF1LL knockdown were due to differential decay, we used REMBRANDTS software, which infers changes in mRNA stability based upon differences in the relative abundance of exonic and intronic reads from each gene (Alkallas et al, 2017). These analyses supported the hypothesis that increases in gene expression upon siUPF1LL were due to inhibition of mRNA decay (FigEV1B and Dataset EV1).

Sashimi plot from representative RNA‐seq samples of siNT and siUPF1LL knockdown cells. Percent spliced in values and FDR were calculated with rMATS software (Shen et al, 2014).
Density plot of changes in mRNA stability as determined by REMBRANDTS analysis of RNA‐seq following isoform‐specific UPF1LL depletion (Alkallas et al, 2017). mRNAs were binned according to up‐ or down‐regulation in response to siUPF1LL. Statistical significance was determined by K–W test, with Dunn's correction for multiple comparisons.
RT–qPCR analysis of indicated transcripts following transfection of HEK‐293 cells with siRNAs that target both UPF1 isoform (UPF1total) and the UPF1LL isoform. Relative fold changes are in reference to NT siRNA. siUPF1total or siUPF1LL was compared to the NT siRNA control for significance testing. Asterisk (*) indicates P
<0.05, as determined by two‐way ANOVA. Black dots represent individual data points and error bars indicate mean±SD (n=3 biological replicates). Dashed lines indicate log2 (fold change) of ±0.5. See also Dataset EV3 for P‐values associated with each statistical comparison.Venn diagram (to scale) of overlapping targets identified from RNA‐seq following UPF1LL knockdown (this dataset), total UPF1 knockdown, or SMG6/7 double knockdown and rescue (Colombo et al, 2017). Depicted are genes that increased in abundance at least 1.4‐fold (FDR
<0.05) with UPF1LL‐specific knockdown and their overlap with genes that increased in abundance (FDR<0.05) with total UPF1 knockdown or genes that increased in abundance with SMG6/7 double knockdown and were significantly rescued by expression of SMG6 or SMG7 (SMG6/7 targets). P‐values indicate enrichment of genes that increased in abundance at least 1.4‐fold (FDR<0.05) with UPF1LL‐specific knockdown among those regulated by total UPF1 and SMG6/7, as determined by Fisher's exact test. Only genes that met read count cutoffs in all conditions were included in the analysis.Density plot of changes in relative mRNA abundance as determined by RNA‐seq in SMG7ko/SMG5kd cells, relative to a parental cell line treated with control siRNAs (Boehm et al, 2021). Genes were categorized as up‐regulated by siUPF1total only, siUPF1LL only, or both siUPF1total and siUPF1LL. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
Density plot of changes in relative mRNA abundance as determined by RNA‐seq following UPF1LL knockdown in HEK‐293 cells. Genes were categorized as up‐regulated by SMG7ko, SMG7ko/SMG5kd, SMG7ko/SMG6kd, or SMG7ko/SMG5kd and SMG7ko/SMG6kd (Boehm et al, 2021). Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
Box plot of log2 enrichment for translation at the ER (Jan et al, 2014). mRNAs were binned by sensitivity to UPF1LL‐specific knockdown in HEK‐293 cells. Statistical significance was determined by K–S test (****P
=1×10−6). Boxes indicate interquartile ranges, horizontal lines represent medians, and bars indicate Tukey whiskers.
Source data are available online for this figure.
To investigate how UPF1LL contributes to overall UPF1 activities in HEK‐293 cells, we compared the transcriptome‐wide effects of specific UPF1LL depletion and knockdown with a pan‐isoform UPF1 siRNA (siUPF1total). Of 1,540 genes that were up‐regulated at least 1.4‐fold upon siUPF1LL treatment and met read count cutoffs in all conditions, 512 overlapped with the 1854 genes more highly expressed upon siUPF1total treatment (Fig1B (bottom) and Dataset EV2). These data suggest that the UPF1LL isoform substantially contributes to UPF1 activities in HEK‐293 cells, despite the UPF1LL mRNA representing only ~15% of UPF1 mRNA in these cells (FigEV1A).
UPF1LL contributes to NMD under normal cellular conditions
We expected that only a subset of genes induced by siUPF1total would be affected by UPF1LL, a prediction borne out by the 1,342 genes (72%) that were uniquely up‐regulated in the siUPF1total condition. However, we also found that 1,028 genes were up‐regulated more than 1.4‐fold upon siUPF1LL but not siUPF1total treatment (Fig1B). Because the existence of a large class of genes regulated by siUPF1LL but not siUPF1total was unexpected, we pursued three strategies to evaluate whether these genes represent genuine siUPF1LL targets: (i) investigation of NMD autoregulation in siUPF1LL and siUPF1total conditions, (ii) comparison to additional published NMD RNA‐seq datasets, and (iii) analysis of mRNA abundance from cells depleted of UPF1total, UPF1LL, or SMG6 by RT–qPCR.
The NMD pathway is governed by a conserved autoregulatory program in which depletion or inactivation of NMD pathway components drives elevated expression of several core NMD factors. NMD feedback regulation has been shown to heavily depend on long 3’UTR‐dependent turnover of NMD factor mRNAs (Singh et al, 2008; Huang et al, 2011; Yepiskoposyan et al, 2011). In siUPF1total RNA‐seq, we observed pathway‐wide induction of NMD factor mRNAs, with expression of mRNAs encoding SMG1, SMG6, SMG7, SMG9, UPF2, UPF3A, and UPF3B all increased at least 1.4‐fold (Fig1C). In contrast, UPF1LL depletion had a minimal impact on NMD factor mRNA expression, failing to perturb any by more than 1.2‐fold. In an independent experiment, we knocked down UPF1total or UPF1LL and evaluated NMD factor mRNAs by RT–qPCR, again observing up‐regulation of NMD factor mRNAs upon siUPF1total but not siUPF1LL treatment (FigEV1C and Dataset EV3). The finding that UPF1total but not UPF1LL depletion induced compensatory up‐regulation of NMD components provides a mechanism to explain why some mRNAs might be de‐repressed by UPF1LL knockdown (which does not induce compensatory feedback regulation of NMD) but not by UPF1total knockdown (which induces up‐regulation of several core NMD factors).
To further evaluate the contribution of UPF1LL to cellular NMD, we compared our UPF1LL‐knockdown RNA‐seq dataset with a published catalog of high‐confidence NMD targets (Colombo et al, 2017). Consistent with the overlap between siUPF1LL and siUPF1total in our RNA‐seq studies, we observed significant overlaps among the population of genes induced by UPF1LL depletion and those previously determined to be repressed by UPF1, SMG6, or SMG7 (FigEV1D), with 618 of the putative UPF1LL targets represented in the published NMD target catalog.
Co‐regulation of UPF1LL target mRNAs by SMG6
To gain insight into the involvement of other NMD pathway components in UPF1LL‐dependent regulation, we took advantage of a recently published RNA‐seq dataset from experiments in which the Gehring laboratory combined CRISPR‐mediated SMG7 knockouts with RNAi‐mediated SMG5 or SMG6 knockdowns (Boehm et al, 2021). For these analyses, we categorized genes as up‐regulated by siUPF1total only, siUPF1LL only, or both siUPF1total and siUPF1LL. All three classes exhibited significantly enhanced expression in SMG7ko/SMG6kd cells, relative to a parental cell line treated with control siRNAs (Fig1D). The greatest degree of up‐regulation in SMG7ko/SMG6kd cells was observed for genes induced by both siUPF1total and siUPF1LL. Genes that responded to only siUPF1LL were up‐regulated in SMG7ko/SMG6kd cells to a very similar extent to those that responded to only siUPF1total.
Interestingly, genes induced by siUPF1LL but not siUPF1total exhibited distinct responses to SMG5 depletion in the SMG7ko background. In contrast to the systematic up‐regulation of the siUPF1LL‐only class of genes in SMG7ko/SMG6kd cells, this group of genes was not on average induced in SMG7ko/SMG5kd cells (FigEV1E). Reciprocally, genes that were up‐regulated in SMG7ko/SMG6kd but not SMG7ko/SMG5kd cells were most substantially induced by UPF1LL depletion (FigEV1F). Together, these analyses support the idea that many genes uniquely up‐regulated by siUPF1LL are genuine NMD pathway targets, as they are responsive to co‐inactivation of NMD factors SMG6 and SMG7. Moreover, these data suggest that these genes, as a class, are particularly dependent on SMG6 for proper regulation.
To corroborate the results of our own and published RNA‐seq datasets, we selected representative genes for evaluation by RT–qPCR of mRNA from cells depleted of UPF1total, UPF1LL, or SMG6 (Fig1E and Dataset EV3). We analyzed genes from three major categories: (i) well‐characterized NMD targets, including EJC‐stimulated alternative splice isoforms of SRSF2, SRSF3, and SRSF6 and long 3’UTR decay targets SMG1 and SMG5, (ii) putative UPF1LL targets regulated by both UPF1LL and UPF1total depletion in our RNA‐seq studies, and (iii) putative UPF1LL targets up‐regulated by UPF1LL depletion but not UPF1total depletion. Knockdown of UPF1LL had no effect on the levels of well‐characterized premature termination codon (PTC)‐containing SRSF2 and SRSF6 transcripts, and increased SRSF3 PTC transcript levels to a much smaller extent (~1.9‐fold) than total UPF1 (~6.2‐fold) or SMG6 (~8.1‐fold) knockdown (Lareau et al, 2007; Ni et al, 2007). Transcriptome‐wide, we found a similar pattern, as depletion of UPF1total but not UPF1LL caused systematically elevated expression of PTC‐containing transcript isoforms relative to control PTC‐free isoforms (Appendix Fig S1C). Importantly, all selected UPF1LL target mRNAs, irrespective of siUPF1total responsiveness, were significantly up‐regulated by SMG6 depletion (Fig1E). These data further reinforce the conclusion that genes responding to UPF1LL but not UPF1total were likely up‐regulated due to UPF1LL depletion rather than off‐target effects.
Targets of UPF1LL are enriched for ER‐associated gene products
The set of genes that respond to siUPF1LL but not siUPF1total is an interesting class because their regulation can be unambiguously attributed to UPF1LL and they can be studied in the absence of the overall NMD pathway up‐regulation that results from knockdown of total UPF1 and other NMD factors. However, it is important to note that we do not currently know the mechanisms that determine whether mRNAs regulated by UPF1LL are responsive to siUPF1LL alone or both siUPF1LL and siUPF1total, as transcripts uniquely affected by siUPF1LL did not show any significant enrichment for specific NMD‐inducing features like PTCs, uORFs, or long 3’UTRs (Appendix TableS1). Therefore, except where noted, further analyses treat the entire population of siUPF1LL‐responsive genes as putative UPF1LL targets, irrespective of the effects of UPF1total knockdown.
Because genes in functionally related pathways are often coordinately regulated at the posttranscriptional level (Keene, 2007), we performed a gene ontology (GO) enrichment analysis (Eden et al, 2009) to identify commonalities among UPF1LL targets. This analysis revealed a high degree of enrichment among UPF1LL targets for genes encoding proteins that rely on the endoplasmic reticulum (ER) for biogenesis (Fig1F and Dataset EV4). In contrast, GO analysis of mRNAs up‐regulated by siUPF1total treatment yielded no significantly enriched categories. In total, 768 of the 1,621 genes up‐regulated by UPF1LL depletion are annotated by UniProt as encoding integral membrane, secreted, and/or signal peptide‐containing proteins. We also used a previous survey of ER‐localized translation (Jan et al, 2014) to corroborate the results of the GO analysis, finding that many UPF1LL target mRNAs were indeed found to be preferentially translated at the ER (FigEV1G).
Affinity purification reveals transcriptome‐wide UPF1SL and UPF1LL binding profiles
The observation that specific depletion of UPF1LL affected a select subpopulation of NMD targets indicated it has distinct cellular functions from those of the major UPF1SL isoform. To gain insight into how the biochemical properties of the two UPF1 isoforms differ, we performed affinity purification followed by RNA‐seq (RIP‐seq) of each UPF1 variant (Fig2A). For these studies, we engineered HEK‐293 stable cell lines to inducibly express CLIP‐tagged UPF1LL or UPF1SL, with a GFP‐expressing stable line as a control, a system that leads to 5‐ to 6‐fold overexpression relative to endogenous UPF1total levels (FigEV2A). We elected to express CLIP‐tagged UPF1 constructs, as the CLIP tag can be covalently biotinylated for efficient isolation by streptavidin affinity purification (Gautier et al, 2008). We have previously used this system to show that biotinylated CLIP‐UPF1SL isolated from human cells preferentially associates with NMD‐susceptible mRNA isoforms (Kishor et al, 2020). Analysis of well‐characterized NMD substrate levels following knockdown of total endogenous UPF1 and rescue with the siRNA‐resistant CLIP‐UPF1 constructs confirmed that both CLIP‐tagged UPF1 isoforms were equally able to function in NMD (FigEV2B).

Scheme for the CLIP‐UPF1 affinity purification (RIP) assay.
Scatterplot of CLIP‐UPF1LL vs. CLIP‐UPF1SL RIP‐seq efficiency. mRNAs were binned according to 3’UTR length (short, medium, or long).
Density plot of recovered mRNAs in CLIP‐UPF1LL or CLIP‐UPF1SL affinity purifications. mRNAs were binned according to 3’UTR length. Statistical significance was determined by K–S test.
Density plot of recovered mRNAs in CLIP‐UPF1LL affinity purifications relative to that of CLIP‐UPF1SL. mRNAs were subdivided by PTBP1 and/or hnRNP L motif density within the 3’UTR, as indicated by the gradient triangle. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
RT–qPCR analysis of indicated transcripts from CLIP‐UPF1 overexpression RNA‐seq experiments. Relative fold changes are in reference to the GFP‐expressing control line. Significance of CLIP‐UPF1SL or CLIP‐UPF1LL overexpression was compared to the GFP‐expressing control line. Asterisk (*) indicates P
<0.05, as determined by two‐way ANOVA. Black dots represent individual data points and error bars indicate mean±SD (n=3 biological replicates). Dashed lines indicate log2 (fold change) of ±0.5. For protected mRNAs, the motif density of PTBP1/hnRNP L within the 3'UTR is indicated. PTC+ indicates the use of primers specific to transcript isoforms with validated poison exons (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P values associated with each statistical comparison.
Source data are available online for this figure.

Western blots of CLIP‐UPF1SL and CLIP‐UPF1LL overexpression. Membranes were probed with an anti‐UPF1 antibody that detects both endogenous and CLIP‐tagged UPF1. Wedge indicates serial twofold dilutions of lysate. Mean (±
SD) of CLIP‐UPF1 overexpression was determined from two replicate membranes.RT–qPCR analysis of well‐characterized NMD targets following total UPF1 knockdown and rescue with siRNA‐resistant CLIP‐tagged UPF1. Relative fold changes are in reference to the GFP‐expressing control line treated with a NT siRNA. Significance of NMD rescue by CLIP‐UPF1 was compared to the GFP‐expressing control line treated with total UPF1 siRNA. Asterisk (*) indicates P
<0.0001, as determined by two‐way ANOVA with multiple comparisons. Black dots represent individual data points and error bars indicate mean±SD (n=3 biological replicates). PTC+ indicates the use of primers specific to transcript isoforms with validated poison exons (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P‐values associated with each statistical comparison.Density plot of recovered mRNAs in CLIP‐UPF1LL affinity purifications relative to that of CLIP‐UPF1SL. Genes were categorized as up‐regulated by siUPF1total only, siUPF1LL only, or both siUPF1total and siUPF1LL. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
RT–qPCR analysis of indicated transcripts from UPF1 RIP‐seq experiments. Relative fold enrichment was determined by dividing the recovered mRNA by its corresponding input amount. Significance of differential recovery in CLIP‐UPF1LL RIP was determined by comparison to that in CLIP‐UPF1SL. Asterisk (*) indicates P
<0.05, as determined by unpaired Student’s t‐test. Black dots represent individual data points, and error bars indicate mean±SD (n=3 biological replicates). For protected mRNAs, the PTBP1/hnRNP L motif density bin of the 3'UTR is indicated. PTC+ indicates the use of primers specific to transcript isoforms with validated poison exons (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P‐values associated with each statistical comparison.Density plots of changes in relative mRNA abundance as determined by RNA‐seq following UPF1LL (top) or UPF1SL (bottom) overexpression. mRNAs were binned according to enrichment in the CLIP‐UPF1LL or CLIP‐UPF1SL affinity purifications. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
Density plots of changes in mRNA stability as determined by REMBRANDTS analysis of RNA‐seq following UPF1LL (top) or UPF1SL (bottom) overexpression. mRNAs were binned according to enrichment in the CLIP‐UPF1LL or CLIP‐UPF1SL affinity purifications. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
Source data are available online for this figure.
CLIP‐UPF1 complexes were isolated from whole cell extracts by streptavidin affinity purification using a CLIP‐biotin substrate, with GFP‐expressing cell lines as a negative control for interaction specificity (Appendix Fig S2A). Bound RNAs were then extracted and used for sequencing library preparation. Because recovery of RNA from GFP samples was at least 100‐fold lower than from CLIP‐UPF1 affinity purifications (Dataset EV5), only UPF1 samples were analyzed by RNA‐seq. UPF1 occupancy was assessed by normalizing the abundance of transcripts in RIP‐seq samples to their abundance in total RNA‐seq (hereafter referred to as UPF1 RIP‐seq efficiency).
The UPF1SL and UPF1LL isoforms differ only in the domain 1B regulatory loop and therefore share a common RNA binding interface composed of residues from the RecA, 1B, 1C, and stalk domains (Chakrabarti et al, 2011). The majority of UPF1‐RNA contacts involve sugar‐phosphate recognition, enabling high‐affinity, sequence‐nonspecific RNA binding. Consistent with these structural features, CLIP‐UPF1SL and CLIP‐UPF1LL exhibited equivalent binding to the vast majority of endogenous mRNAs (mRNAs from 9711 of 10,673 genes were recovered within ±1.4‐fold in the two conditions; Fig2B and Dataset EV5).
UPF1LL preferentially associates with long 3’UTRs
UPF1 accumulates in a length‐dependent manner on 3’UTRs due to its active displacement from coding regions by translating ribosomes and its nonspecific RNA binding activity (Hogg & Goff, 2010; Hurt et al, 2013; Zünd et al, 2013; Kurosaki et al, 2014; Baker & Hogg, 2017). To evaluate the relationship between 3’UTR length and CLIP‐UPF1 binding, we determined the distribution of RIP‐sequencing efficiencies in three 3’UTR length bins (first tertile: <566nt; second tertile: 566–1,686nt; third tertile: >1,686nt). Consistent with previous findings, the efficiency of mRNA co‐purification with both UPF1SL and UPF1LL increased with 3’UTR length (Fig2C).
A potential caveat to the RIP‐seq studies is that the CLIP‐tagged UPF1 proteins are ~5 to 6‐fold overexpressed relative to endogenous UPF1total (FigEV2A), which may impair the assay’s discriminative power between the two isoforms. In line with this idea, binding of CLIP‐UPF1LL and CLIP‐UPF1SL to mRNAs from genes induced upon siUPF1LL and siUPF1total treatment was equivalent in these assays (FigEV2C). However, one class of transcript, those with long 3’UTRs, was more efficiently co‐purified with CLIP‐UPF1LL than CLIP‐UPF1SL (Fig2B and C). We therefore asked whether this preferential enrichment may give clues to distinct biochemical properties of the two isoforms.
Enhanced UPF1LL binding to NMD‐resistant transcripts
Transcripts with long 3’UTRs represent a large population of potential NMD targets (Yepiskoposyan et al, 2011; Hurt et al, 2013), only some of which are degraded by the pathway under normal conditions (Toma et al, 2015). Providing a biochemical mechanism to explain evasion of long 3’UTRs from decay, we have identified hundreds to thousands of mRNAs shielded by the protective RNA‐binding proteins (RBPs) PTBP1 and hnRNP L (Ge et al, 2016; Kishor et al, 2019b, 2020). In our previous work, we showed that increased PTBP1 and/or hnRNP L motif binding density within the 3’UTR correlates with reduced UPF1SL binding and recovery of mRNAs in UPF1SL RIP‐seq studies (Ge et al, 2016; Kishor et al, 2019b; Fritz etal, 2020). Based on the observation that UPF1LL more efficiently recovers the longest class of 3’UTRs, we asked whether mRNAs protected by PTBP1 and/or hnRNP L are differentially associated with UPF1LL versus UPF1SL.
Subdivision of the transcriptome first by 3’UTR length and then according to the density of PTBP1 and hnRNP L binding sites within the 3’UTR revealed that transcripts with long 3’UTRs and moderate or high densities of protective protein binding sites were more efficiently recovered by CLIP‐UPF1LL than CLIP‐UPF1SL (Fig2D and Appendix Fig S2B). This preferential recovery of long 3’UTRs with moderate or high densities of protective protein binding by CLIP‐UPF1LL was similarly observed when PTBP1 and/or hnRNP L motif densities were restricted to the first 400nt of the 3’UTR (Appendix Fig S2C), which we previously established as a strong feature driving protection and reduced UPF1SL binding (Ge et al, 2016; Kishor et al, 2019b). Quantitative RT–PCR of select transcripts confirmed these transcriptome‐wide RIP‐seq results (FigEV2D and Dataset EV3).
If UPF1LL can more efficiently associate with mRNAs normally shielded from NMD by the protective RBPs, then we would expect a correlation between the transcripts affected by protective protein depletion and those enriched for UPF1LL binding. Indeed, mRNAs preferentially recovered by CLIP‐UPF1LL were significantly down‐regulated in response to PTBP1 depletion in HEK‐293 cells from our previous work (Ge et al, 2016; Data ref: Ge et al, 2016), a result expected for NMD substrates normally shielded by the protective RBP (Appendix Fig S2D). We observed a similar downregulation of mRNAs enriched for CLIP‐UPF1LL binding in a publicly available RNA‐seq dataset of mouse neuronal progenitor cells depleted of PTBP1 and its brain‐specific paralogue PTBP2 (Appendix Fig S2E; Linares et al, 2015; Data ref: Linares et al, 2015). Together, our findings indicate that the distinct biochemical properties of UPF1LL give it the capacity to circumvent PTBP1 and/or hnRNP L to associate with otherwise protected mRNAs.
UPF1LL overexpression down‐regulates mRNAs normally protected from NMD
We further analyzed the RNA‐seq data from RIP‐seq input samples to ask whether differential transcript recognition by the UPF1 isoforms was reflected in differential regulation upon CLIP‐UPF1SL or CLIP‐UPF1LL overexpression. mRNAs preferentially bound by CLIP‐UPF1LL were systematically down‐regulated upon CLIP‐UPF1LL overexpression, but not CLIP‐UPF1SL overexpression (FigEV2E, top). Conversely, a small population of mRNAs preferentially recovered by CLIP‐UPF1SL were down‐regulated by CLIP‐UPF1SL but not CLIP‐UPF1LL overexpression (FigEV2E, bottom). To investigate whether the observed changes in mRNA abundance with UPF1 overexpression were due to enhanced decay, we again used REMBRANDTS software (Alkallas et al, 2017). These analyses found that regulation of gene expression upon UPF1 overexpression was attributable to decreased mRNA stability (FigEV2F and Dataset EV5).
We additionally corroborated the transcriptome‐wide results obtained using RNA‐seq by performing RT–qPCR on select transcripts (Fig2E and Dataset EV3). Notably, validated mRNAs down‐regulated by UPF1LL overexpression include CSRP1, which we have previously established as a long 3’UTR‐containing mRNA that undergoes decay upon hnRNP L knockdown or mutation of hnRNP L binding sites in its 3’UTR (Kishor et al, 2019b). Reduced exogenous expression of CLIP‐UPF1LL to levels ~0.7‐fold that of total endogenous UPF1 (Appendix Fig S3A) had only small effects on levels of protected mRNAs (Appendix Fig S3B), indicating that removal of protection requires a more substantial perturbation of UPF1LL expression. Together, these data support the conclusion that the UPF1LL isoform is biochemically equipped to overcome the protective proteins to promote decay of mRNAs normally shielded from NMD, but that in cells with normal endogenous UPF1SL levels, protection is maintained unless UPF1LL is substantially overexpressed.
SRSF1 is required for expression of the UPF1LL splice isoform
Knockdown and overexpression of UPF1LL involve drastic changes in UPF1LL abundance, both in absolute terms and relative to UPF1SL. We reasoned that manipulation of a regulator of UPF1 alternative splicing might allow us to manipulate the UPF1LL:UPF1SL ratio without changing the total cellular UPF1 expression level. We surveyed publicly available alternative splicing analysis from ENCODE RBP knockdown RNA‐seq data, which reported reduced UPF1LL splice isoform selection upon knockdown of the serine/arginine‐rich splicing factor 1 (SRSF1) in K562 and HepG2 cells (Yee et al, 2019; Van Nostrand et al, 2020). Consistent with ENCODE data, we observed substantial and specific loss of the UPF1LL mRNA isoform in semi‐quantitative RT–PCR assays from HEK‐293 cells treated with SRSF1 siRNAs (Fig3A). To test the functional effects of SRSF1‐mediated UPF1 splicing regulation, we depleted SRSF1 from cells overexpressing CLIP‐UPF1SL or CLIP‐UPF1LL. We chose to focus on CSRP1 mRNAs for these experiments because we have extensively validated the role of hnRNP L in antagonizing NMD of these transcripts (Kishor et al, 2019b) and, in this study, have shown that they are down‐regulated by CLIP‐UPF1LL overexpression (Fig2E). Knockdown of SRSF1 caused increased CSRP1 mRNA expression, an effect that was reversed by overexpression of CLIP‐UPF1LL but not CLIP‐UPF1SL (Fig3B and Dataset EV3).

Semiquantitative RT–PCR of UPF1SL or UPF1LL transcript levels following transfection of HEK‐293 cells with the indicated siRNAs.
RT–qPCR analysis of indicated transcripts following transfection of a NT siRNA or SRSF1‐specific siRNA under conditions of CLIP‐UPF1SL or CLIP‐UPF1LL overexpression. Relative fold changes are in reference to the GFP‐expressing control line treated with a NT siRNA. Asterisk (*) indicates P
<0.05, as determined by unpaired Student’s t‐test. Black dots represent individual data points and error bars indicate mean±SD (n=3 biological replicates). See also Dataset EV3 for Pvalues associated with each statistical comparison.Density plot of changes in relative mRNA abundance as determined by RNA‐seq following SRSF1 overexpression (Data ref: Caputi et al, 2019). Genes were categorized as up‐regulated by siUPF1total only, siUPF1LL only, or both siUPF1total and siUPF1LL. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.
Density plot as in (C), with genes binned according to enrichment in the CLIP‐UPF1LL or CLIP‐UPF1SL affinity purifications.
Source data are available online for this figure.
We next asked whether SRSF1 overexpression would enhance use of the UPF1LL isoform and, if so, whether an elevated UPF1LL:UPF1SL ratio would affect transcripts we have identified as responsive to UPF1LL knockdown or overexpression. We analyzed a public RNA‐seq dataset from cells in which SRSF1 was overexpressed (Data ref: Caputi et al, 2019), finding an ~1.6‐fold increase in usage of the UPF1LL mRNA isoform in SRSF1 overexpression relative to vector control cells (SRSF1 overexpression Ѱ=27.8%; vector control Ѱ=17.7%; Appendix Fig S4). This increase in UPF1LL:UPF1SL ratio was associated with decreased expression of mRNAs identified as up‐regulated in our siUPF1LL RNA‐seq dataset, while the expression of mRNAs up‐regulated by only siUPF1total was decreased (Fig3C). Correspondingly, mRNAs preferentially bound by CLIP‐UPF1LL versus CLIP‐UPF1SL were systematically down‐regulated by SRSF1 overexpression (Fig3D). Together, these data establish SRSF1 as a regulator of UPF1 alternative splicing. Moreover, they indicate that relatively subtle changes in UPF1LL:UPF1SL isoform ratio are sufficient to significantly favor or impair UPF1LL activities in cells.
UPF1LL is less sensitive to PTBP1‐mediated inhibition of translocation
We have proposed that the protective RBPs PTBP1 and hnRNP L exploit the tendency of UPF1 to release RNA upon ATP binding and hydrolysis to promote UPF1 dissociation from potential NMD substrates prior to decay induction (Fritz et al, 2020). In support of this model, deletion of the regulatory loop, which mediates ATPase‐dependent dissociation, rendered UPF1SL less sensitive to PTBP1 inhibition in vitro (Fritz et al, 2020). Importantly, both the physiological UPF1LL isoform and the engineered UPF1 variant containing a regulatory loop deletion exhibit a greater affinity for RNA in the presence of ATP than the PTBP1‐sensitive UPF1SL isoform (Gowravaram et al, 2018). We therefore hypothesized that UPF1LL can mimic the ability of the loop truncation mutant to overcome negative regulation by PTBP1.
We recently established a real‐time assay to monitor UPF1 translocation activity (Fritz et al, 2020). In this assay, UPF1 translocation and duplex unwinding causes a fluorescently labeled oligonucleotide to be displaced from the assay substrate (Fig4A, left). An excess of complementary oligonucleotide labeled with a dark quencher is provided in the reaction, causing a decrease in fluorescence with increased displacement of the labeled oligonucleotide by UPF1. Inhibition of UPF1 translocation results in sustained fluorescence over time, allowing for the determination of inhibitory effects of PTBP1 on UPF1 unwinding activity. Using this assay in our previous work, we obtained evidence that PTBP1 inhibits UPF1 translocation rather than initial binding (Fritz et al, 2020). This inhibitory effect on UPF1 translocation activity was specific to PTBP1 and was not observed in the presence of the high‐affinity RNA binding Pseudomonas phage 7 coat protein, supporting the conclusions that the protective proteins specifically promote the dissociation of UPF1 and that our assay can robustly assess inhibitors of UPF1 unwinding activity.

Scheme of the fluorescence‐based unwinding assay to monitor UPF1 translocation in real‐time (Fritz et al, 2020). An RNA substrate harboring a high‐affinity PTBP1 binding site is incubated with highly purified UPF1ΔCH in the absence and presence of equal molar amounts of highly purified PTBP1. Upon the addition of ATP, UPF1 translocation results in a decrease in fluorescence due to displacement of a labeled, duplexed oligonucleotide and subsequent quenching by a trap strand.
UPF1SLΔCH translocation along an RNA substrate harboring a high‐affinity PTBP1 binding site in the absence and presence of PTBP1. Time to 50% unwound and relative total % unwound at end of assay (1,200
s) are indicated. Results of four technical replicates are shown for each dataset and represent at least three independent experiments. Shaded region indicates SD.Results as in (B) but with UPF1LLΔCH.
Source data are available online for this figure.
We therefore leveraged this system to compare UPF1LL versus UPF1SL translocation on a duplexed RNA substrate harboring a high‐affinity PTBP1 binding site (Fig4A, right) (Fritz et al, 2020). For these experiments, we compared the activity of highly purified UPF1 proteins containing the helicase core but lacking the autoinhibitory N‐terminal cysteine‐histidine domain (UPF1ΔCH) (Appendix Fig S5A and B; Chakrabarti et al, 2011; Fiorini et al, 2012; Fritz et al, 2020). UPF1SLΔCH exhibited robust unwinding activity in the absence of PTBP1, displacing 50% of the duplexed oligonucleotide in 100s (Fig4B). This translocation activity of UPF1SLΔCH was dependent upon the addition of ATP, as previously demonstrated (Fritz et al, 2020). Addition of PTBP1 substantially impaired UPF1SLΔCH unwinding activity, reducing both the rate at which the oligonucleotide was displaced (requiring 360s to attain the half‐maximal unwinding value reached by UPF1SLΔCH alone) and the overall extent of unwinding (73% of the UPF1SLΔCH total at the end of the assay).
UPF1LLΔCH also exhibited robust unwinding activity in the absence of PTBP1, displacing 50% of the duplexed oligonucleotide in 90s in an ATP‐dependent manner (Fig4C). The observed enhancement in UPF1LLΔCH translocation activity over UPF1SLΔCH is consistent with previous reports of increased catalytic activity of the UPF1LL isoform relative to UPF1SL (Gowravaram et al, 2018). In contrast to UPF1SLΔCH, UPF1LLΔCH maintained robust unwinding activity in the presence of PTBP1, displacing 50% of the duplexed oligonucleotide by 180s and achieving 94% total duplex unwinding at the end of the assay. These results indicate that UPF1LL can overcome the translocation inhibition by PTBP1, reinforcing the conclusion that PTBP1‐mediated UPF1 inhibition depends on the clash between the UPF1 regulatory loop and RNA.
Coordinated downregulation of UPF1LL targets during ER stress and ISR induction
Our in vitro, RIP‐seq, and overexpression studies suggested that UPF1LL has the biochemical capacity to expand the scope of UPF1‐dependent regulation. Based on these observations, we next investigated whether specific physiological conditions might promote changes in NMD target susceptibility by harnessing endogenous UPF1LL activity. Multiple lines of evidence led us to examine the regulation of UPF1LL in the integrated stress response (ISR), which restores homeostasis by repressing translation and inducing expression of a battery of stress response genes (Fig5A; Costa‐Mattioli & Walter, 2020). Activation of the ISR induces hyperphosphorylation of eIF2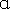 , driving global downregulation of protein synthesis due to impaired eIF2‐GTP‐Met‐tRNAi ternary complex recycling and reduced delivery of the initiator Met‐tRNAi to translating ribosomes (Baird & Wek, 2012; Young & Wek, 2016; Wek, 2018). An established effect of ISR‐mediated translational repression is corresponding stabilization of well‐characterized NMD targets, including several mRNAs encoding factors integral to the activation and resolution of the stress response (Goetz & Wilkinson, 2017).
, driving global downregulation of protein synthesis due to impaired eIF2‐GTP‐Met‐tRNAi ternary complex recycling and reduced delivery of the initiator Met‐tRNAi to translating ribosomes (Baird & Wek, 2012; Young & Wek, 2016; Wek, 2018). An established effect of ISR‐mediated translational repression is corresponding stabilization of well‐characterized NMD targets, including several mRNAs encoding factors integral to the activation and resolution of the stress response (Goetz & Wilkinson, 2017).

Scheme for activation of the integrated stress response (ISR) and effects on UPF1‐dependent decay.
Density plots of changes in relative mRNA abundance as determined by RNA‐seq following treatment of HEK‐293 cells with 1
µM thapsigargin for 6h (left) or 9h (right). Genes were categorized as up‐regulated by siUPF1total only or siUPF1LL only under basal conditions. Statistical significance was determined by K–W test, with Dunn’s correction for multiple comparisons.RNA‐seq analysis of HEK‐293 cells identifies populations of genes that decreased in abundance with thapsigargin treatment and were rescued by UPF1LL‐specific knockdown. Indicated are genes that increased in abundance at least 1.4‐fold (FDR
<0.05) with UPF1LL‐specific knockdown under normal conditions.RT–qPCR analysis of indicated transcripts following transfection of HEK‐293 cells with indicated siRNAs and treatment with 1
µM thapsigargin for 6h. Relative fold changes are in reference to vehicle‐treated, NT siRNA. Black dots represent individual data points and error bars indicate mean±SD (n=3 biological replicates). Dashed lines indicate log2 (fold change) of ±0.5. PTC+ indicates the use of primers specific to transcript isoforms with validated poison exons (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P‐values associated with each statistical comparison.
Source data are available online for this figure.
Two of our findings led us to consider the possibility that UPF1LL activity is regulated by the ISR. First, GO analyses indicated substantial enrichment of ER‐localized mRNAs among those up‐regulated by UPF1LL knockdown (Fig1F). Second, as an initial test of the hypothesis that certain cellular conditions would promote turnover of mRNAs preferentially bound by CLIP‐UPF1LL, we analyzed a published RNA‐seq dataset from cells treated with the ISR‐inducing agent tunicamycin (Park et al, 2017; Data ref: Park et al, 2017). This analysis identified systematic downregulation of mRNAs enriched in CLIP‐UPF1LL RIP‐seq, in contrast to RNAs preferentially bound by CLIP‐UPF1SL (FigEV3A).

Volcano plot of relative mRNA abundance as determined from RNA‐seq following treatment of HEK‐293 cells with 1
µM tunicamycin for 6h (Data ref: Park et al, 2017). mRNAs were binned by RIP‐seq efficiency in CLIP‐UPF1LL or CLIP‐UPF1SL affinity purifications. Statistical significance was determined by K–W test, with Dunn's correction for multiple comparisons. Dashed line indicates the significance threshold P≤0.05 (n=3 biological replicates).Western blot of eIF2α phosphorylation following treatment of HEK‐293 cells with 1
µM thapsigargin for 6h.Schematic of the RNA‐seq experimental workflow and conditions for UPF1LL knockdown and thapsigargin treatment.
Density plot of relative mRNA stability as determined by REMBRANDTS analysis of RNA‐seq following treatment of HEK‐293 cells with 1
µM thapsigargin for 6h. mRNAs were binned by changes in relative mRNA abundance in thapsigargin. Statistical significance was determined by K–S test.Density plot of relative mRNA stability as determined by REMBRANDTS analysis of RNA‐seq following UPF1LL knockdown in HEK‐293 cells and treatment with 1
µM thapsigargin for 6h (Alkallas et al, 2017). mRNAs were binned by changes in relative mRNA abundance in thapsigargin with UPF1LL knockdown. Statistical significance was determined by K–W test, with Dunn's correction for multiple comparisons.Quantification of characterized ISR‐target transcript abundance in RNA‐seq of the indicated conditions. Error bars indicate mean
±SD (n=3 biological replicates).Quantification of UPF1LL isoform expression in control and thapsigargin‐treated HEK‐293 cells from rMATS analyses (n
=3 biological replicates).
Source data are available online for this figure.
We next directly assessed how UPF1LL activity contributes to gene expression regulation during ER stress and induction of the ISR by performing RNA‐seq of HEK‐293 cells treated with the ER stress‐inducing agent thapsigargin (Fig5B and Dataset EV6). Western blot analysis showed a 2.5‐fold increase in eIF2 phosphorylation with thapsigargin treatment (FigEV3B), supporting a robust induction of the ISR. Consistent with previous results (Nickless et al, 2014; Li etal, 2017), mRNAs up‐regulated upon total UPF1 knockdown in HEK‐293 cells were on average also up‐regulated following 6 or 9h in 1μM thapsigargin (Fig5B), and the magnitude of the increase correlated with the effects of UPF1 total knockdown. In sharp contrast, genes up‐regulated by UPF1LL‐specific knockdown exhibited a distinct behavior upon thapsigargin treatment. Rather than increasing, UPF1LL substrates showed on average a reduction in mRNA levels, and this tendency did not vary according to the magnitude of the effect of UPF1LL knockdown. These results indicate that UPF1LL functions distinctly from that of well‐characterized NMD and sustains activity during ER stress and activation of the ISR.
UPF1LL conditionally remodels NMD target selection during ER stress and ISR induction
To more comprehensively evaluate the role of UPF1LL in promoting the downregulation of select genes during ISR induction, we transfected HEK‐293 cells with non‐targeting (NT) or UPF1LL‐specific siRNAs and then treated cells with 1μM thapsigargin for 6 or 9h (FigEV3C). In RNA‐seq analyses, we identified 606 genes that significantly decreased in abundance with thapsigargin treatment, of which 135 (6h) or 143 (9h) were rescued at least 1.4‐fold upon UPF1LL knockdown (Fig5C, Appendix Fig S6A, and Dataset EV6). In contrast, only 70 (6h) or 62 (9h) of these 606 genes decreased in abundance in response to UPF1LL knockdown in thapsigargin treatment. These results were highly reproducible between the 6h and 9h thapsigargin RNA‐seq datasets (Appendix Fig S6B), supporting that a unique population of genes are selectively down‐regulated during conditions of ER stress and induction of the ISR in a UPF1LL‐dependent manner.
Inferred mRNA stability changes using REMBRANDTS software supported that the observed differences in mRNA abundance upon thapsigargin treatment and UPF1LL knockdown were due to changes in mRNA decay (FigEV3D and E). The changes in gene expression caused by UPF1LL depletion were not attributable to differential ISR induction, as previously established stress response genes were comparably up‐regulated in response to thapsigargin treatment following NT and UPF1LL‐specific knockdown (FigEV3F; Ashburner et al, 2000; The Gene Ontology Consortium, 2019). Moreover, thapsigargin treatment did not alter the relative levels of UPF1LL and UPF1SL mRNAs (FigEV3G), indicating that UPF1LL activity in ER stress was likely due to activity of the existing population of protein rather than a consequence of altered UPF1 splicing upon thapsigargin treatment.
Our finding that UPF1LL has the potential to bind and regulate transcripts protected from NMD by PTBP1 and/or hnRNP L under normal cell growth conditions (Figs2 and EV2) led us to ask whether genes down‐regulated by UPF1LL during ISR induction included substrates beyond those identified as UPF1LL targets under normal cellular conditions (Fig1). Of the 135 genes down‐regulated by UPF1LL upon thapsigargin treatment, 49 genes (36%) were unique to the population of UPF1LL targets down‐regulated during ISR induction, while 86 genes were identified as UPF1LL targets under both normal and stress conditions (Fig5C). These data indicate that UPF1LL activity is maintained or enhanced when cells are subjected to ER stress conditions that inhibit well‐characterized NMD events.
To corroborate the above findings, we performed RT–qPCR on select transcripts identified by RNA‐seq as constitutively or conditionally regulated by UPF1LL (Fig5D and Dataset EV3). As in the RNA‐seq analyses, RT–qPCR of putative condition‐specific UPF1LL targets revealed several mRNAs (e.g., TMEM165, LDLR, TIMP2, NXT2, TGOLN2, and COL4A1) that exhibited UPF1LL‐dependent downregulation upon thapsigargin treatment but were not affected by siUPF1LL under normal growth conditions. Taken together, these data support a model in which expression of dual UPF1SL and UPF1LL isoforms enable conditional remodeling of NMD target selection in response to ISR induction.
UPF1LL target repertoire is expanded by partial translationalrepression
Because NMD requires detection of in‐frame stop codons, target susceptibility is sensitive to changes in the location and frequency of translation initiation and termination. In addition to modulation of initiation via eIF2 phosphorylation in ER stress (Goetz & Wilkinson, 2017), inhibition of translation elongation (e.g., with cycloheximide and puromycin) inhibits the decay of well‐characterized NMD targets (Carter et al, 1995). We therefore asked whether translational repression would promote UPF1LL activity outside of the context of the ISR (Fig6A).

Framework for investigation of effects of translation inhibition on UPF1LL activity.
RNA‐seq analysis of HEK‐293 cells identifies populations of genes that decreased in abundance with puromycin treatment (50
µg/ml for 4h) and were rescued by UPF1LL‐specific knockdown. Indicated are genes that increased in abundance at least 1.4‐fold (FDR<0.05) with UPF1LL‐specific knockdown under normal conditions.RT–qPCR analysis of indicated transcripts following transfection of HEK‐293 cells with indicated siRNAs and treatment with 50
µg/ml puromycin for 4h. Relative fold changes are in reference to vehicle‐treated, NT siRNA. Black dots represent individual data points, and error bars indicate mean±SD (n=3 biological replicates). Dashed lines indicate log2 (fold change) of ±0.5. PTC+ indicates the use of primers specific to transcript isoforms with validated poison exons (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P‐values associated with each statistical comparison.
Source data are available online for this figure.
We again compared published RNA‐seq datasets with our UPF1 RIP‐seq data to provide a preliminary test of this hypothesis. As we observed using published RNA‐seq data from cells undergoing ER stress, mRNAs preferentially bound by UPF1LL were systematically down‐regulated upon treatment with moderate doses of the initiation inhibitor hippuristanol or the elongation inhibitors emetine and cycloheximide (FigEV4A–C) (Data ref: Martinez‐Nunez & Sanford, 2016; Martinez‐Nunez et al, 2017; Kearse et al, 2019; Data ref: Kearse et al, 2019; Waldron et al, 2019; Data ref: Waldron et al, 2019). In the case of cycloheximide treatment, only 15min of treatment was sufficient to observe significant loss of UPF1LL‐enriched transcripts, consistent with a decay‐driven process.

Volcano plot of relative mRNA abundance as determined from RNA‐seq following treatment of MCF7 cells with 150
nM hippuristanol for 1h (Data ref: Waldron et al, 2019). mRNAs were binned by RIP‐seq efficiency in CLIP‐UPF1LL or CLIP‐UPF1SL affinity purifications. Statistical significance was determined by K–W test, with Dunn's correction for multiple comparisons. Dashed line indicates the significance threshold P≤0.05 (n=3 biological replicates).Volcano plot as in (A), following treatment of HEK‐293 cells with 50
ng/ml emetine for 4h (Martinez‐Nunez et al, 2017). Dashed line indicates the significance threshold P≤0.05 (n=3 biological replicates).Volcano plot as in (A), following treatment of HeLa cells with 100
µg/ml cycloheximide for 15min (Data ref: Kearse et al, 2019). Dashed line indicates the significance threshold P≤0.05 (n=3 biological replicates).
We next used siUPF1LL treatment to directly test whether mRNAs down‐regulated upon moderate translation inhibition required UPF1LL. We elected to use the translation elongation inhibitor puromycin for these experiments because it is widely used to inhibit canonical NMD events and acts through a completely distinct mechanism from the block to initiation caused by eIF2 phosphorylation. Specifically, the ribosome catalyzes the linkage of puromycin to nascent polypeptides, causing chain termination and peptide release (Nathans, 1964).
To investigate whether UPF1LL is able to exert sustained or even enhanced post‐transcriptional control in response to translation inhibition via distinct mechanisms, we transfected HEK‐293 cells with NT or UPF1LL‐specific siRNAs and then treated cells with puromycin (50µg/ml for 4h; FigEV5A). Remarkably, RNA‐seq analyses revealed 2,279 genes that significantly decreased in abundance with puromycin treatment, of which 700 (31%) were rescued at least 1.4‐fold upon UPF1LL knockdown (Fig6B and Dataset EV7). In contrast, only 124 genes (5%) decreased in abundance in response to UPF1LL knockdown in puromycin treatment (Appendix Fig S7A). The response to UPF1LL depletion was highly consistent in cells treated with in 25, 50, and 100µg/ml puromycin (Appendix Fig S7B), supporting a role for UPF1LL in gene expression regulation during conditions of partial translational repression. Genes identified as destabilized in REMBRANDTS analysis of CLIP‐UPF1LL versus CLIP‐UPF1SL overexpression tended to be down‐regulated upon puromycin treatment, while the reverse was true for genes preferentially destabilized by CLIP‐UPF1SL overexpression (FigEV5B). Consistent with a specific role for UPF1LL, knockdown of UPF1LL ameliorated the effects of puromycin treatment on genes regulated by CLIP‐UPF1LL but not CLIP‐UPF1SL overexpression.

Schematic of the RNA‐seq experimental workflow and conditions for UPF1LL knockdown and puromycin treatment.
Density plot of relative mRNA abundance as determined by RNA‐seq following treatment of HEK‐293 cells with 50
µg/ml puromycin. mRNAs were binned according to destabilization in CLIP‐UPF1LL or CLIP‐UPF1SL overexpression experiments, as determined by REMBRANDTS analysis (Alkallas et al, 2017). Statistical significance was determined by K–S test.Quantification of UPF1LL isoform expression in control and puromycin‐treated HEK‐293 cells from rMATS analyses (n
=3 biological replicates) (Shen et al, 2014).RT–qPCR analysis of indicated transcripts following treatment of HEK‐293 cells with indicated concentrations of puromycin for 4
h. Relative fold changes are in reference to vehicle‐treated control. Significance of puromycin treatment on relative transcript abundance was compared to the vehicle‐treated control. Asterisk (*) indicates P<0.05, as determined by two‐way ANOVA. Black dots represent individual data points, and error bars indicate mean±SD (n=3 biological replicates). Dashed lines indicate log2 (fold change) of ±0.5. PTC+ indicates the use of primers specific to the transcript isoform with a validated poison exon (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P‐values associated with each statistical comparison.Density plot of relative mRNA abundance as determined by RNA‐seq following treatment of HEK‐293 cells with 25
µg/ml or 100µg/ml puromycin. mRNAs were binned according to sensitivity to 50µg/ml puromycin and UPF1LL knockdown. Statistical significance was determined by K–S test.Density plot of relative mRNA stability as determined by REMBRANDTS analysis of RNA‐seq following treatment of HEK‐293 cells with 50
µg/ml puromycin for 4h (Alkallas et al, 2017). mRNAs were binned by changes in relative mRNA abundance in puromycin. Statistical significance was determined by K–S test.Density plot of relative mRNA stability as determined by REMBRANDTS analysis of RNA‐seq following UPF1LL knockdown in HEK‐293 cells and treatment with 50
µg/ml puromycin for 4h (Alkallas et al, 2017). mRNAs were binned by changes in relative mRNA abundance in puromycin with UPF1LL knockdown. Statistical significance was determined by K–W test, with Dunn's correction for multiple comparisons.
Source data are available online for this figure.
We next employed puromycin to identify new conditional UPF1LL targets, finding that 550 genes (79%) identified as rescued by UPF1LL from puromycin‐dependent downregulation were uniquely affected during puromycin treatment. The remaining 150 genes (21%) significantly overlapped with those up‐regulated with UPF1LL depletion under normal cellular conditions (Fig6B). RT–qPCR of select transcripts confirmed the transcriptome‐wide RNA‐seq results (Fig6C and Dataset EV3). Similar to conditions of ER stress, puromycin treatment did not alter UPF1 splicing (FigEV5C), indicating that UPF1LL activity during conditions of impaired translation was likely due to the existing population of UPF1LL protein. These data support that translational repression promotes UPF1LL activity outside of the context of the ISR to conditionally remodel NMD target selection.
UPF1LL activity requires ongoing translation
Because of the well‐established requirement for translation in NMD, we hypothesized that the UPF1 isoform‐dependent effects of thapsigargin and moderate puromycin treatment were due to changes in the location and/or frequency of translation termination events (Fig6A). To test this hypothesis, we treated cells with a titration of puromycin from 25µg/mL to 400µg/ml. If UPF1LL activity depends on the infrequent residual translation termination events that occur under puromycin treatment, its activity should be enhanced at low concentrations of puromycin that permit some termination events to persist but be inhibited by high concentrations of puromycin that more efficiently block translation.
In line with these expectations, we observed a dose‐dependent response, in which downregulation of representative UPF1LL target transcripts FMR1, eIF5A2, ERBB2, and CDK16 was most efficient at lower puromycin concentrations (FigEV5D and Dataset EV3). Treatment with high concentrations of puromycin did not have a significant effect on the levels of these UPF1LL target mRNAs, consistent with a requirement for translation termination events. Corroborating these findings, we observed globally more efficient downregulation of puromycin‐sensitive UPF1LL targets with 25µg/ml puromycin than 100µg/ml puromycin in RNA‐seq (FigEV5E and Dataset EV7). Based on these results, we conclude that translation termination is likely required for all UPF1‐dependent decay events but that changes in translation efficiency can drive the downregulation of a novel class of substrates by the UPF1LL isoform.
Translational repression promotes UPF1LL‐dependent decay of select mRNAs
Inferred mRNA stability changes using REMBRANDTS software indicated that the observed differences in mRNA abundance upon puromycin treatment and UPF1LL knockdown from the RNA‐seq studies were due to corresponding changes in mRNA stability (FigEV5F and G and Dataset EV7). To directly evaluate the effect of translational repression on promoting the decay of mRNAs by UPF1LL, we leveraged the recently established method of Roadblock‐qPCR to assess endogenous mRNA stability (Watson et al, 2021). In this method, 4‐thiouridine (4‐SU) is used to label transcripts produced during a 4‐h timecourse. Isolated RNA is treated with N‐ethylmaleimide, which covalently labels 4‐SU residues, forming a bulky adduct that blocks reverse transcription. RT–qPCR of the remaining unlabeled pool thus allows straightforward quantification of mRNA turnover.
HEK‐293 cells were transfected with NT or UPF1LL‐specific siRNAs and labeled with 4‐SU in the absence and presence of puromycin. In this analysis, puromycin treatment stabilized the canonical NMD target of ATF4 (Fig7A and Dataset EV3), consistent with previous findings that translational repression inhibits decay of well‐characterized NMD targets. In contrast, representative UPF1LL targets CDK16 and TNFRSF10D exhibited significantly shorter half‐lives with puromycin treatment, an effect that was dependent upon UPF1LL expression. Together, these data support the conclusion that translational repression promotes the decay of mRNAs by UPF1LL.

mRNA decay measurement using Roadblock‐qPCR (Watson et al, 2021). RNA was isolated from HEK‐293 cells at indicated timepoints following transfection with indicated siRNAs and treatment with 400
µM 4sU and 50µg/ml puromycin. mRNA half‐lives were estimated by fitting the data to a single‐phase exponential decay model. Puromycin treatment was compared to the vehicle control and siUPF1LL was compared to the siNT in the absence and presence of puromycin treatment using the extra‐sum‐of‐squares F test. Error bars indicate mean±SD (n=4 biological replicates). See also Dataset EV3 for P values associated with each statistical comparison.RT–qPCR analysis of indicated transcripts following transfection of HEK‐293 cells with indicated siRNAs and treatment with 50
µg/ml puromycin for 4h. Relative fold changes are in reference to vehicle‐treated, NT siRNA. Black dots represent individual data points and error bars indicate mean±SD (n=3 biological replicates). Dashed lines indicate log2 (fold change) of ±0.5. PTC+ indicates the use of primers specific to transcript isoforms with validated poison exons (Lareau et al, 2007; Ni et al, 2007). See also Dataset EV3 for P values associated with each statistical comparison.Density plot of changes in relative mRNA abundance as determined from RNA‐seq following treatment of HEK‐293 cells with 50
µg/ml of puromycin for 4h. mRNAs were subdivided by PTBP1 and/or hnRNP L motif density within the first 400nt of 3'UTR. Statistical significance was determined by K–W test, with Dunn's correction for multiple comparisons.Density plot as in (C), following UPF1LL‐specific knockdown.
Source data are available online for this figure.
UPF1LL activity during translational repression requires SMG6
We next asked whether UPF1LL‐dependent mRNA downregulation during translational repression involved the specialized NMD endonuclease SMG6. To explore this possibility, HEK‐293 cells were transfected with NT or SMG6‐specific siRNAs and then treated with puromycin. Quantitative RT–PCR of select transcripts revealed that knockdown of SMG6 significantly rescued the downregulation of UPF1LL targets in puromycin treatment (Fig7B and Dataset EV3). This effect was comparable to that observed with UPF1LL‐specific depletion, supporting the conclusion that select mRNA downregulation during translational repression is due to UPF1LL activity in the NMD pathway. Furthermore, knockdown of SMG6 under normal conditions increased the abundance of well‐characterized NMD targets and constitutively regulated UPF1LL substrates but did not significantly affect the levels of conditionally regulated UPF1LL substrates. These results provide further evidence that UPF1LL remodels NMD target selection during translational repression to promote the decay of a new class of substrates.
NMD‐protected mRNAs are down‐regulated by UPF1LL during translational repression
Finally, we asked whether the expanded functions of UPF1LL in translational repression are related to its biochemical capability to direct degradation of mRNAs normally protected by PTBP1 and/or hnRNP L. We analyzed whether RNAs with stop codon‐proximal PTBP1 and hnRNP L motifs are among those susceptible to UPF1LL‐mediated downregulation upon puromycin treatment. Subdivision of the transcriptome according to PTBP1 and/or hnRNP L motif binding density within the first 400nt of the 3’UTR revealed that mRNAs with high densities of binding sites for the protective proteins were significantly down‐regulated with puromycin treatment relative to mRNAs with low densities of binding sites (Fig7C). Knockdown of UPF1LL rescued this decrease in mRNA abundance, supporting the conclusion that the downregulation of protected mRNAs during translational repression was dependent upon UPF1LL expression (Fig7D). Based on these data, we conclude that enhanced UPF1LL activities upon translational repression result in deprotection of normally NMD‐insensitive mRNAs.
Discussion
Here, we employ specific depletion, overexpression, and biochemical methods to identify that the mammalian UPF1LL isoform performs distinct functions from that of the major UPF1SL isoform. By depleting only the UPF1LL‐encoding mRNA, we show that UPF1LL is required for a subset of UPF1‐mediated regulation, preferentially targeting mRNAs that encode transmembrane and secreted proteins translated at the ER (Fig1). Our transcriptome‐wide studies of UPF1LL and UPF1SL RNA binding reveal that UPF1LL has a greater capacity to bind and regulate mRNAs normally protected from decay by PTBP1 and hnRNP L (Fig2). The endogenous expression of UPF1LL depends on the splicing regulator SRSF1; correspondingly, manipulation of SRSF1 levels by knockdown or overexpression causes impaired or enhanced UPF1LL activity, respectively (Fig3). The cellular interaction specificity of UPF1LL is corroborated by its ability to overcome inhibition by PTBP1 in vitro (Fig4), consistent with our previous observation that PTBP1 promotes ATPase‐dependent UPF1 dissociation by exploiting the UPF1 regulatory loop (Fritz et al, 2020). These data in sum suggest that UPF1LL has the biochemical capability to regulate the protected population of mRNAs but that its activities are likely constrained by its relatively low expression in HEK‐293 and many other cell types.
In contrast to the well‐characterized inhibition of NMD by ER stress, we find that UPF1LL‐dependent regulation is intact or even enhanced (Fig5). Mechanistically, preferential UPF1LL activity in ER stress can be explained by its ability to function under conditions of translational repression (Fig6). Moderate inhibition of translation with puromycin causes thousands of genes to be down‐regulated, of which approximately one‐third are rescued by UPF1LL knockdown. These experiments show that UPF1LL is not only required for downregulation of mRNAs identified as substrates under normal conditions (“constitutive” UPF1LL targets), but also regulates additional mRNAs under ER stress and translational repression (“conditional” UPF1LL targets). mRNAs that are down‐regulated by UPF1LL upon puromycin treatment are enriched for stop codon‐proximal protective protein binding sites, providing a mechanism for their conditional, isoform‐specific targeting (Fig7). Combined with the inhibition of UPF1SL‐dependent decay, enhanced UPF1LL activity upon translational repression results in a dramatic and unanticipated shift in NMD target specificity.
We present a model in which moderate translational repression does not uniformly repress NMD but instead alters its specificity (Fig8). In this model, the relative activity of UPF1LL expands as translation activity decreases, allowing continued degradation of constitutive UPF1LL targets and inducing downregulation of new substrate mRNAs. The change in NMD specificity upon translational repression is enabled by the distinct biochemical properties of the UPF1LL protein. First, UPF1LL has an intrinsically enhanced affinity for RNA in the presence of ATP (Gowravaram et al, 2018), which may allow prolonged association with mRNAs and increase the probability that UPF1 binding co‐occurs with translation termination. Second, UPF1LL is able to overcome inhibition by protective proteins such as PTBP1, which allows targeting of mRNAs normally shielded from decay (Fig2).

See Discussion for details.
We envision that the two UPF1 isoforms may compete for less abundant NMD factors, particularly under conditions that impair decay or translation. An attractive possibility is that persistent binding of UPF1LL to mRNAs promotes its phosphorylation by SMG1, readying UPF1LL to scaffold assembly of productive decay complexes (Kurosaki et al, 2014; Durand et al, 2016). In this scenario, mRNAs with a greater relative propensity to bind UPF1LL vs. UPF1SL—those normally shielded by protective proteins—are thus poised to be decayed upon inhibition of translation. A second potential cause of transcript deprotection is that the location of the most frequently used stop codon changes, for example if termination at an unprotected stop codon at the end of an upstream ORF becomes more frequent than termination at a protected stop codon at the end of the main ORF.
This model is built on the fundamental concept that when translation is partially repressed, translation termination events still occur, but their relative frequencies across the transcriptome are expected to be altered. We show that diverse mechanisms of translation inhibition promote UPF1LL activity (Figs5, ,6,6, ,77 and FigEV4A–C), but our model predicts that the precise specificity of decay will be inhibitor‐dependent. For example, the spectrum of termination events that occur upon inhibition of translation initiation is expected to be distinct from those that occur when elongation is blocked. Likewise, distinct cell types and growth conditions will have distinct translation termination landscapes, which will in turn lead to different collections of UPF1LL and UPF1SL targets. From a practical perspective, our data highlight the importance of careful control of cell growth conditions when studying UPF1LL and other aspects of NMD, as changes in translation due to differential nutrient availability, confluence, and other factors can negatively and positively affect decay in a substrate‐specific manner.
The idea that the mammalian NMD pathway consists of multiple branches with distinct factor requirements and substrate specificities has been proposed by several groups, but the underlying mechanisms and regulatory roles of NMD branching are poorly understood (Gehring et al, 2005; Chan et al, 2007; Huang et al, 2011; Yi et al, 2021). Unraveling NMD pathway branches has been challenging, in part because most NMD proteins are essential for cell viability, precluding conclusive demonstrations that they are dispensable for decay of any given NMD target mRNA (Kishor et al, 2019a; Boehm et al, 2021; Yi et al, 2021). Our identification of differential activities of UPF1SL and UPF1LL is an unforeseen example of NMD pathway branching, which can be controlled at the cellular level by changes in the abundance of protective RBPs, translation, or UPF1 splicing. The differential regulation of UPF1LL‐dependent targets in SMG7ko/SMG6kd and SMG7ko/SMG5kd cells provides an initial indication that previously described NMD branches may be distinctly influenced by UPF1LL (Figs1D and EV1E and F; (Boehm et al, 2021).
Because altered translation efficiency and induction of cellular stress pathways are pervasive in cancer, genetic disease, and infection, the expanded scope of UPF1LL‐dependent decay has far‐reaching implications for the physiological roles of the mammalian NMD pathway in health and disease. In particular, the ability of UPF1LL to target specific mRNAs in response to translational repression positions NMD to function as a mechanism to reset the transcriptome upon cellular stress. The extent to which UPF1LL complements or collaborates with IRE1‐mediated mRNA decay or the recently identified ER‐associated NMD factor NBAS will require further study (Hollien & Weissman, 2006; Longman et al, 2020), but our data suggest that UPF1LL may help to relieve proteotoxic stress by reducing the abundance of transcripts encoding proteins with complex biosynthetic needs.
Even in the absence of stress, cells and tissues in vivo likely have lower basal rates of translation than those attained in exponentially growing transformed cell lines. Based on our findings, understanding how NMD shapes gene expression in diverse physiological contexts will require not just analysis of NMD targets characterized in transformed cells, but also transcripts that are conditionally targeted in response to changing translational states. Notably, we find that UPF1LL is able to conditionally regulate several proteins of central importance in cancer and other diseases, including fragile X mental retardation 1 (FMR1), the low‐density lipoprotein receptor (LDLR), and oncogenes PTEN, EIF5A2, and ERBB2, among others (Goldstein & Brown, 2009; Song et al, 2012; Mathews & Hershey, 2015; Dockendorff & Labrador, 2019; Harbeck et al, 2019). Along with EIF5A2 and FMR1, several additional UPF1LL targets that undergo enhanced downregulation upon translational repression are themselves important regulators of translation and mRNA decay, including the signal recognition particle receptor SRPR, the major mRNA decapping enzyme DCP2, and the miRNA‐processing endonuclease DICER1 (Akopian et al, 2013; Mugridge et al, 2018; Michlewski & Cáceres, 2019).
Materials and Methods
UPF1 isoform nomenclature
UPF1SL refers to the protein encoded by Ensembl transcript ID ENST00000262803.9 and UPF1LL by Ensembl transcript ID ENST00000599848.5. These isoforms are also referred to as UPF1 isoform 2 and UPF1 isoform 1, respectively (Nicholson et al, 2014; Gowravaram et al, 2018).
In vitro helicase assays
Unwinding assays were performed as previously described in detail (Fritz et al, 2020). Briefly, 75nM of the pre‐assembled RNA duplex substrate (described below) was combined with 1× unwinding reaction buffer (10mM MES pH 6.0, 50mM KOAc, 0.1mM EDTA), 2mM MgOAc, and 1‐unit RNasin in a well of a half‐area, black, flat‐bottom 96‐well plate (Corning 3993). PTBP1 (80nM) was then added, the sample mixed, and incubated at room temperature for 10min. Then, UPF1 (80nM) was added, the sample mixed, and incubated at room temperature for 10min. Finally, BHQ1 quencher (0.56µM final; GTGTGCGTACAACTAGCT/3BHQ_1) and 2mM ATP were added to initiate the unwinding reaction. An InfiniteR F200 Pro microplate reader and associated i‐controlTM 1.9 software (Tecan) was used to monitor Alexa Fluor 488 fluorescence every 10s for 20min at 37°C. Measured fluorescence intensities were normalized to the zero timepoint for each condition to obtain relative fluorescence values. Four technical replicates for each condition were obtained at the same time. The decrease in fluorescence caused by UPF1 unwinding in the absence of PTBP1 at the end of each 20‐min timecourse was used to calculate the time to 50% maximal unwinding and relative total unwinding for each condition. Because measurements were taken at 10‐s intervals, the earliest time at which 50% maximal unwinding was observed is indicated on each graph.
To generate the RNA duplex, a DNA oligo template (5’ TAATACGACTCACTATAGGGACACAAAACAAAAGACAAAAACACAAAACAAAAGACAAAAACACAAAACAAAAGACAAAAAGCCTCTCCTTCTCTCTGCTTCTCTCTCGCTGTGTGCGTACAACTAGCT 3’) was PCR‐amplified and then in vitro transcribed using the MEGAshortscriptTM T7 Transcription kit (Invitrogen). An 11:7 ratio of the helicase RNA substrate to 5’ Alex Fluor 488 fluorescent oligo strand (Alexa Fluor 488/AGCTAGTTGTACGCACAC) was incubated with 2mM MgOAC and 1× unwinding reaction buffer at 95°C for 3min and 30s and then slowly cooled to 30°C. All DNA oligos were obtained from Integrated DNA Technologies. The 5’ fluorescent oligo probe and 3’ BHQ1 quencher were acquired as RNase‐free, HPLC‐purified.
Cloning, expression, and purification of recombinant UPF1LLΔCH, UPF1SLΔCH, and PTBP1 were conducted as previously described (Gowravaram et al, 2018; Fritz et al, 2020). In this study, UPF1LLΔCH was purified in the same manner as UPF1SLΔCH (Fritz et al, 2020).
Mammalian cell lines and generation of CLIP‐UPF1 expression lines
HEK‐293 cells used in the endogenous UPF1LL knockdown and RNA‐seq experiments were received from ATCC (CRL‐3216) and maintained at 37°C and 5% CO2 in DMEM with 10% FBS (Gibco) and 1% pen/strep. Stable integration of CLIP‐UPF1SL into human Flp‐In™ T‐Rex™‐293 cells (Invitrogen) and subsequent maintenance of this stable line was previously described (Kishor et al, 2020). CLIP‐UPF1LL expression lines were generated and maintained in an identical manner. Total UPF1 and SMG6 depletion studies followed by RT–qPCR as well as the Roadblock‐qPCR experiments were performed using HEK‐293 cells maintained as described above. Total UPF1 depletion and RNA‐seq was conducted using parental Flp‐In™ T‐Rex™‐293 cells (Invitrogen) maintained according to manufacturer instructions. To express CLIP‐UPF1 at levels ~0.7‐fold that of the endogenous protein, the CMV promoter and 5’ UTR hairpin from pcDNA HP40GFPTP (Hogg & Goff, 2010) were inserted in place of the tet‐regulated pFRT/TO promoter in the CLIP‐UPF1 expression plasmid (Kishor et al, 2020) via SpeI and HindIII sites. Stable cell lines Flp‐In™ T‐Rex™‐293 in which the resulting UPF1 expression plasmids were integrated were prepared and maintained as above.
Endogenous UPF1, SMG6, or SRSF1 depletion by siRNA
The following siRNAs were used to deplete indicated UPF1 isoforms, SMG6, or SRSF1: total UPF1 (Forward sequence: 5’ CUACCAGUACCAGAACAUA 3’; Reverse sequence: 5’ UAUGUUCUGGUACUGGUAG 3’); UPF1LL (Forward sequence: 5’ GGUAAUGAGGAUUUAGUCA 3’; Reverse sequence: 5’ UGACUAAAUCCUCAUUACC 3’); SMG6 (Forward sequence: 5’ GCUGCAGGUUACUUACAAG 3’; Reverse sequence: 5’ CUUGUAAGUAACCUGCAGC 3’; Durand et al, 2016); SRSF1 (Forward sequence: 5’ UGAAGCAGGUGAUGUAUGU 3’; Reverse sequence: 5’ ACAUACAUCACCUGCUUCA 3’; (de Miguel et al, 2014)).
CLIP‐UPF1 overexpression RIP‐seq and RNA‐seq
CLIP‐UPF1SL overexpression RIP‐seq and RNA‐seq sample preparation were previously reported (Kishor et al, 2020); CLIP‐UPF1LL datasets were generated in parallel. Briefly, CLIP‐UPF1 stable cell lines or a GFP‐expressing control line were seeded in 6×15cm plates and then treated with 200ng/ml doxycycline hyclate (Sigma) for 48h to induce CLIP‐UPF1 expression. Cells were harvested 48h post‐induction at 80–85% confluency and whole cell lysate generated by the freeze/thaw method as previously described (Hogg & Collins, 2007a, 2007b; Hogg & Goff, 2010; Fritz et al, 2018). Equilibrated cell extracts, reserving 1/10th for downstream input analysis, were then combined with 10µM CLIP‐Biotin (New England Biolabs) and rotated (end‐over‐end) for 1h at 4°C to allow CLIP‐UPF1 to react with the CLIP‐Biotin substrate and yield covalently biotinylated protein. Unbound CLIP‐Biotin was subsequently removed by passing the samples through Zeba™ Spin Desalting Columns, 40K MWCO, 2ml (Thermo Scientific) according to manufacturer instructions. Buffer exchange was performed with HLB‐150 supplemented with 1mM DTT and 0.1% NP‐40. Pre‐washed Dynabeads™ MyOne™ Streptavidin T1 (Invitrogen) were then added, and the samples rotated (end‐over‐end) for 1h at 4°C to immobilize the biotin‐bound CLIP‐UPF1 complexes. The samples were then washed three times with 500 µL of HLB‐150 supplemented with 1mM DTT and 0.1% NP‐40 and 1/100th reserved for downstream Western blot analysis of CLIP‐UPF1 pull‐down efficiency. The remainder was combined with TRIzol™ (Invitrogen) and RNA was isolated according to manufacturer instructions. DNase treatment was subsequently performed using RQ1 RNase‐Free DNase (Promega), and RNA was isolated by acid phenol chloroform extraction. This resulted in approximately 1µg of total RNA that was then subjected to high‐throughput sequencing. In parallel, the reserved input lysate (approximately 3µg of total RNA) was also sent for high‐throughput sequencing. A total of three biological replicates from each condition were processed. Sequencing libraries were prepared from input and bound RNA using the Illumina TruSeq Stranded Total RNA Human kit and sequenced on an Illumina HiSeq 3000 instrument.
To validate select transcripts by RT–qPCR, an equivalent volume of reserved RNA (3µl) was used as a template for cDNA synthesis using the Maxima First Strand cDNA synthesis kit for RT–qPCR (Thermo Scientific) according to manufacturer instructions. The resulting cDNA was diluted with nuclease‐free water and subsequently analyzed by qPCR using iTaq Universal SYBR Green Supermix (Bio‐Rad) on a Roche LightCycler 96 instrument (Roche). Sequences for gene‐specific primers used for amplification are listed in Appendix TableS2. For input samples, relative fold change was determined by calculating 2−ΔΔCT values using GAPDH for normalization. For pull‐down samples, relative fold enrichment was determined by dividing the C
q value of the pull‐down by its corresponding input, multiplying by 100 and then normalizing to the relative recovery of the SMG1 transcript.
For assessment of CLIP‐UPF1 expression and pull‐down efficiency by Western blot, reserved input and IP samples were run on a NuPage™ 4–12% Bis‐Tris Protein Gel (Invitrogen) using MOPS buffer according to manufacturer instructions and subsequently transferred to a nitrocellulose membrane according to the NuPage™ manufacturer's protocol (Invitrogen). Membranes were incubated with a blocking buffer for fluorescent Western blotting (Rockland) for 1h at room temperature and then incubated overnight at 4°C with the indicated primary antibody. Primary antibodies used: anti‐RENT1 (goat polyclonal, Bethyl, A300‐038A, 1:1,000) and anti‐β‐actin (mouse monoclonal, Cell Signaling, #3700, 1:1,000). Membranes were subsequently washed three times with 1×TBS supplemented with 0.1% Tween‐20 and then incubated with the appropriate secondary antibody for 1h at room temperature. Secondary antibodies used: anti‐goat IgG (H&L) Antibody DyLight™ 680 Conjugated (Rockland, 605‐744‐002, 1:10,000) and anti‐mouse IgG (H&L) Antibody DyLight™ 680 Conjugated (Rockland, 610‐744‐124, 1:10,000). Membranes were washed three times with 1×TBS supplemented with 0.1% Tween‐20 and then two times with 1×TBS. Western blot images were obtained on an Amersham Typhoon imaging system (GE Healthcare Life Sciences) and quantified using ImageStudio software (LI‐COR Biosciences).
For total UPF1 or SRSF1 depletion and rescue with CLIP‐UPF1, 3×105 cells from the CLIP‐UPF1 stable cell lines, which were engineered as resistant to the described total UPF1 siRNA, or the GFP‐expressing control line were reverse transfected with 40nM of a NT, pan‐UPF1, or SRSF1‐specific siRNA (described above) using Lipofectamine RNAiMAX according to manufacturer instructions. The next day, cells were treated with 200ng/ml doxycycline hyclate (Sigma) for 48h to induce expression of CLIP‐UPF1. Cells were harvested 48h post‐induction, and total RNA was isolated using TRIzol™ (Invitrogen) according to manufacturer instructions. DNase treatment was subsequently performed using RQ1 RNase‐Free DNase (Promega), and RNA was isolated by acid phenol chloroform extraction according to standard protocol. RT–qPCR was performed as described above.
Total UPF1 depletion and RNA‐seq
3×105 HEK‐293 cells (Invitrogen) were reverse transfected with 40nM of a NT or UPF1‐specific siRNA that targets both UPF1 isoforms (described above) using Lipofectamine RNAiMAX according to manufacturer instructions. Seventy‐two hours post‐siRNA transfection, total RNA was isolated using TRIzol™ (Invitrogen) according to manufacturer instructions. DNase‐treatment was subsequently performed using RQ1 RNase‐Free DNase (Promega), and RNA was isolated by acid phenol chloroform extraction according to standard protocol. A total of 2µg RNA was then subjected to high‐throughput sequencing. Three replicates were processed for each condition. Sequencing libraries were prepared using the Illumina TruSeq Stranded Total RNA Human kit and sequenced on an Illumina NovaSeq 6000 instrument.
Total UPF1 and SMG6 depletion with RT–qPCR
3×105 HEK‐293 cells were reverse transfected with 40nM of a NT, total UPF1, or SMG6‐specific siRNA (described above) using Lipofectamine RNAiMAX according to manufacturer instructions. Forty‐eight hours post‐siRNA transfection, cells were replated at a density of 5×105 cells per well of a 6‐well plate. This step was critical to achieve 70–75% confluency on the day of drug treatment (if applicable) and cell harvest. The next day, cells were directly harvested or treated with 50µg/ml puromycin (Sigma) for 4h. A total of three replicates were generated for each condition. Total RNA was then isolated using TRIzol™ (Invitrogen) according to manufacturer instructions. DNase treatment was subsequently performed using RQ1 RNase‐Free DNase (Promega), and RNA was isolated by acid phenol chloroform extraction according to standard protocol. For RT–qPCR, 500ng of total RNA was used as input for cDNA synthesis using the Maxima First Strand cDNA synthesis kit for RT–qPCR (Thermo Scientific) according to manufacturer instructions. The resulting cDNA was diluted with nuclease‐free water and subsequently analyzed by qPCR using iTaq Universal SYBR Green Supermix (Bio‐Rad) on a Roche LightCycler 96 instrument (Roche). Sequences for gene‐specific primers used for amplification are listed in Appendix TableS2. Relative fold changes were determined by calculating 2−ΔΔCT values using GAPDH for normalization.
Endogenous UPF1LL knockdown with puromycin or thapsigargin treatment and RNA‐seq
3×105 HEK‐293 cells were reverse transfected with 40nM of a NT or UPF1LL‐specific siRNA (described above) using Lipofectamine RNAiMAX according to manufacturer instructions. Forty‐eight hours post‐siRNA transfection, cells were replated at a density of 5×105 cells per well of a 6‐well plate. The next day, cells were treated with vehicle control, 25, 50, or 100µg/ml puromycin (Sigma) for 4h, or 1µM thapsigargin for 6 or 9h. A total of three replicates were generated for each condition. Total RNA was then isolated using the RNeasy Plus Mini Kit (QIAGEN). Sequencing libraries were prepared from 2µg total RNA using the Illumina TruSeq Stranded Total RNA Human kit and sequenced on an NovaSeq 6000 instrument. For RT–qPCR validation of changes in target gene expression, 500ng of total RNA was used as input for cDNA synthesis using the Maxima First Strand cDNA synthesis kit for RT–qPCR (Thermo Scientific) according to manufacturer instructions. The resulting cDNA was diluted with nuclease‐free water and subsequently analyzed by qPCR using iTaq Universal SYBR Green Supermix (Bio‐Rad) on a Roche LightCycler 96 instrument (Roche). Sequences for gene‐specific primers used for amplification are listed in Appendix TableS2. Relative fold changes were determined by calculating 2−ΔΔCT values using GAPDH for normalization.
Roadblock‐qPCR to measure endogenous mRNA stability
mRNA decay measurements were determined using Roadblock‐qPCR as previously described (Watson et al, 2021) but with the following adaptations. 3×105 HEK‐293 cells were reverse transfected with 40nM of a NT or UPF1LL‐specific siRNA (described above) using Lipofectamine RNAiMAX according to manufacturer instructions. Forty‐eight hours post‐siRNA transfection, cells were replated at a density of 5×105 cells per well of a 6‐well plate. The next day, cells were treated with 400µM 4‐thiouridine (4sU; Cayman Chemical) and vehicle control or 50µg/ml puromycin (Sigma) for a total of 4h. Cells were harvested at indicated timepoints and total RNA was isolated using TRIzolTM (Invitrogen) according to manufacturer instructions, but with the addition of 1mM (final) DTT to the isopropanol precipitation in order to maintain 4sU in a reduced state (Schofield et al, 2018). Isolated RNA from 4sU‐exposed cells was treated with 48mM N‐ethylmaleimide (NEM; Sigma) as described by Watson et al and then purified using RNAClean XP beads (Beckman Coulter) according to manufacturer instructions. A total of 1µg RNA was used as input for cDNA synthesis with oligo dT18 primers and Protoscript II reverse transcriptase (New England BioLabs) according to manufacturer instructions. The resulting cDNA was diluted with nuclease‐free water and subsequently analyzed by qPCR using iTaq Universal SYBR Green Supermix (Bio‐Rad) on a Roche LightCycler 96 instrument (Roche). Sequences for gene‐specific primers used for amplification are listed in Appendix TableS2. Relative fold changes were determined by calculating 2−ΔΔCT values using time 0 as the reference and GAPDH for normalization. mRNA half‐lives were estimated by fitting the data to a single‐phase exponential decay model using GraphPad Prism 9.1.0.
Western for phospho‐eIF2
HEK‐293 cells treated with 1µM thapsigargin were lysed in 1X Passive Lysis Buffer (Promega) supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific) according to manufacturer instructions. A total of 5µg protein was run on a NuPage™ 4–12% Bis‐Tris Protein Gel (Invitrogen) using MOPS buffer according to manufacturer instructions and subsequently transferred to a nitrocellulose membrane according to the NuPage™ manufacturer's protocol (Invitrogen). Detection of phospho and total eIF2 was performed as previously described (Young‐Baird et al, 2020).
Semiquantitative PCR to detect UPF1 isoform ratios
Generated cDNA (1/40th) from RNA‐seq samples was used as input for PCR amplification with Phusion High‐Fidelity DNA polymerase (New England Biolabs) and UPF1‐specific primers that flank the regulatory loop sequence (Forward: 5’ AACAAGCTGGAGGAGCTGTGGA 3’; Reverse: 5’ ACTTCCACACAAAATCCACCTGGAAGTT 3’). The PCR cycling conditions used were an initial denaturation at 98°C for 30s and then 22 cycles of 98°C for 10s, 63°C for 30s, and 72°C for 15s. PCR products were then run on a 8% Novex™ TBE gel (Invitrogen) according to manufacturer instructions and subsequently stained with SYBR® Gold Nucleic Acid Stain (Invitrogen). Images were obtained on an Amersham Typhoon imaging system (GE Healthcare Life Sciences) and quantified using ImageStudio software (LI‐COR Biosciences).
Gene‐level differential expression analysis
For analysis of RNA‐seq data from HEK‐293 cells treated with NT, anti‐UPF1total, or anti‐UPF1LL siRNAs, raw fastq reads from the NovaSeq 6000 platform were trimmed with fastp (Chen et al, 2018), with the parameters ‐‐detect_adapter_for_pe and ‐‐trim_poly_g. Trimmed reads were aligned with HISAT2 to the hg19/GRCh37 genome and transcriptome index provided by the authors (Kim et al, 2019). For gene‐level differential expression analysis of in‐house and published datasets, reads mapping to Ensembl GRCh37 release 75 gene annotations were quantified with featureCounts (Liao et al, 2014), and differential gene expression was analyzed using limma/voom, as implemented by the Degust server (Powell, 2015; Ritchie et al, 2015). GO analysis was performed with DAVID Bioinformatics Resources, using UniProt keyword classifiers and genes that were represented by >0.5 transcripts per million (TPM) in the RNA‐seq datasets as a background set (Huang et al, 2009a, 2009b; UniProt Consortium, 2021). All comparisons among datasets were performed using genes with average TPM >0.5 in all datasets. Analysis of data from (Boehm et al, 2021) was performed using the authors’ analysis of RNA‐seq of SMG7ko line 34; similar results were observed with SMG7ko line 2.
Isoform‐level differential expression analysis
For isoform‐level differential expression analysis, trimmed reads were quantified against a custom HEK‐293 transcriptome index, prepared with Stringtie and TACO as described (Pertea et al, 2015; Niknafs et al, 2017; Kishor et al, 2019b), using kallisto software with parameters ‐‐bias ‐b 1000 ‐t 16 ‐‐single ‐‐rf‐stranded ‐l 200 ‐s 20 (Bray et al, 2016). Differential transcript expression analysis was performed using RUVSeq and edgeR (Robinson et al, 2009; Risso et al, 2014). Normalization and batch correction were performed with the RUVg function, based on transcripts with invariant expression among all samples. The edgeR TMM method was used to obtain normalized differential expression values and to calculate FDRs. Differential isoform usage was calculated for the most abundant PTC and non‐PTC isoforms of each gene using IsoformSwitchAnalyzer and the DEXSeq package (Anders et al, 2012; Vitting‐Seerup & Sandelin, 2019). Alternative splicing was analyzed using rMATS 4.0.1 (Shen et al, 2014), and Sashimi plots were generated using Integrative Genomics Viewer 2.8.2 (Thorvaldsdóttir et al, 2013). ENCODE rMATS data were downloaded from: https://github.com/YeoLab/rbp‐maps (Yee et al, 2019).
Analysis of mRNA features
IsoformSwitchAnalyzeR was used to annotate PTCs in the custom HEK‐293 transcriptome, as described (Kishor et al, 2019b; Vitting‐Seerup & Sandelin, 2019). For PTC analysis, IsoformSwitchAnalyzeR orfMethod “longest” setting was used to predict the longest ORF (min. 100nt) in each annotated transcript, and TCs located >50nt upstream of the final exon junction were designated potential PTCs. Genes represented by at least one transcript predicted to contain a TC within 50nt of the final exon junction or in the last exon and at least one transcript predicted to contain a PTC, defined as a TC more than 50nt upstream of the final exon junction, were selected for analysis of differential isoform usage upon total UPF1 and UPF1LL‐specific knockdown in Appendix Fig S1C. Putative uORFs were identified using the IsoformSwitchAnalyzeR orfMethod “mostUpstream” setting (min. length 60nt). ORFs predicted by this method that were upstream of the longest predicted ORF were designated potential uORFs. For Appendix TableS1, genes were assigned as PTC‐ or uORF‐containing if at least one transcript with >0.5 TPM in all siUPF1total, siUPF1LL, and siNT samples was found to have a putative PTC or uORF.
The most abundant transcript isoform from each gene, as determined by quantification with kallisto as above, was used for analysis of 3’UTR length and PTBP1 and hnRNP L binding motif positions and frequencies. PTBP1 and hnRNP L binding motif position‐specific scoring matrices were downloaded from the RBPmap database and used for motif finding in 3’UTRs derived from the custom HEK‐293 transcriptome with HOMER, as described (Heinz et al, 2010; Paz et al, 2014; Kishor et al, 2019b).
RIP‐seq analysis
Raw fastq reads from CLIP‐UPF1 overexpression RNA‐seq and RIP‐seq data were trimmed with Cutadapt using the following parameters: ‐‐times 2 ‐e 0 ‐O 5 ‐‐quality‐cutoff 6 ‐m 18 ‐a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC ‐A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT ‐b AAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA ‐b TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT (Martin, 2011). Trimmed reads were quantified using kallisto software with parameters ‐‐bias ‐b 1000 ‐t 16 ‐single ‐‐rf‐stranded ‐l 200 ‐s 20 (Bray et al, 2016). RIP‐seq enrichment values were obtained by dividing TPM values from IP samples by TPM values from input samples.
GTEx data analysis
GTEx data were downloaded from the GTEx Portal on 11/11/2020. To determine the relative representation of UPF1LL and UPF1SL mRNA isoforms, transcript TPM values for transcript ENST00000599848.5 (UPF1LL) were divided by the total TPM values derived from transcripts ENST00000599848.5 (UPF1LL) and ENST00000262803.9 (UPF1SL). GTEx samples were assigned to the indicated tissue types using the sample attributes provided in GTEx Analysis v8.
Statistical analysis
Statistics and exponential decay fits were calculated using GraphPad Prism 9. Density plots were generated using the JMP 14 (SAS Institute) One‐way Platform, with default values. All statistical tests were two‐sided, and all replicates shown from cell‐based experiments are biological replicates, defined as experiments performed with independently manipulated cell populations. For helicase assays, biological replicates are defined as biochemical assays performed with independent reaction mixtures at different times and technical replicates are defined as reactions performed at the same time. Where indicated in the figure legends, P‐values are reported in Dataset EV3.
Author contributions
Sarah E Fritz: Conceptualization; Data curation; Formal analysis; Investigation; Visualization; Writing—original draft; Writing—review and editing. Soumya Ranganathan: Investigation; Methodology; Writing—review and editing. Clara D Wang: Investigation; Writing—review and editing. J Robert Hogg: Conceptualization; Data curation; Formal analysis; Supervision; Funding acquisition; Investigation; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
SEF and JRH conceived and designed the study, analyzed and interpreted the data, and wrote the manuscript. SEF acquired the UPF1 RIP‐seq and RNA‐seq data and performed associated cellular studies. SR acquired the unwinding data. CDW performed the total UPF1 knockdown for RNA‐seq. All authors read and approved the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Sutapa Chakrabarti for the UPF1LLΔCH recombinant expression plasmid and members of the Hogg laboratory and Nicholas R. Guydosh for critical reading of the manuscript. We are grateful to Sandy Mattijssen and Richard J. Maraia for helpful discussion and to Sara K. Young‐Baird for thapsigargin reagents, helpful discussion, and assistance with phospho‐eIF2 immunoblotting. High‐throughput sequencing was conducted by Yan Luo and Poching Liu in the NHLBI DNA Sequencing and Genomics Core. The Genotype‐Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. This work was supported by the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute and utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Notes
The EMBO Journal (2022) 41: e108898. [Europe PMC free article] [Abstract] [Google Scholar]
Data availability
- • RNA‐Seq data: Gene Expression Omnibus GSE134059 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134059).
- • RNA‐Seq data: Gene Expression Omnibus GSE162699 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162699).
- • RNA‐Seq data: Gene Expression Omnibus GSE176197 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE176197).
References
- Akopian D, Shen K, Zhang X, Shan S‐O (2013) Signal recognition particle: an essential protein‐targeting machine. Annu Rev Biochem 82: 693–721 [Europe PMC free article] [Abstract] [Google Scholar]
- Alkallas R, Fish L, Goodarzi H, Najafabadi HS (2017) Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer’s disease. Nat Commun 8: 1–11 [Europe PMC free article] [Abstract] [Google Scholar]
- Anders S, Reyes A, Huber W (2012) Detecting differential usage of exons from RNA‐seq data. Genome Res 22: 2008–2017 [Europe PMC free article] [Abstract] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [Europe PMC free article] [Abstract] [Google Scholar]
- Baird TD, Wek RC (2012) Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 3: 307–321 [Europe PMC free article] [Abstract] [Google Scholar]
- Baker SL, Hogg JR (2017) A system for coordinated analysis of translational readthrough and nonsense‐mediated mRNA decay. PLoS One 12: e0173980 [Europe PMC free article] [Abstract] [Google Scholar]
- Boehm V, Kueckelmann S, Gerbracht JV, Kallabis S, Britto‐Borges T, Altmüller J, Krüger M, Dieterich C, Gehring NH (2021) SMG5‐SMG7 authorize nonsense‐mediated mRNA decay by enabling SMG6 endonucleolytic activity. Nat Commun 12: 1–19 [Europe PMC free article] [Abstract] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 34: 525–527 [Abstract] [Google Scholar]
- Caputi M, Clark E, Paz S (2019) Gene Expression Omnibus GSE124397 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124397). [DATASET]
- Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF (1995) A regulatory mechanism that detects premature nonsense codons in T‐cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro . J Biol Chem 270: 28995–29003 [Abstract] [Google Scholar]
- Causier B, Li Z, De Smet R, Lloyd JPB, Van de Peer Y, Davies B (2017) Conservation of nonsense‐mediated mRNA decay complex components throughout eukaryotic evolution. Sci Rep 7: 1–12 [Europe PMC free article] [Abstract] [Google Scholar]
- Chakrabarti S, Jayachandran U, Bonneau F, Fiorini F, Basquin C, Domcke S, Le Hir H, Conti E (2011) Molecular mechanisms for the RNA‐dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell 41: 693–703 [Abstract] [Google Scholar]
- Chamieh H, Ballut L, Bonneau F, Le Hir H (2008) NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol 15: 85–93 [Abstract] [Google Scholar]
- Chan W‐K, Huang L, Gudikote JP, Chang Y‐F, Imam JS, MacLean JA II, Wilkinson MF (2007) An alternative branch of the nonsense‐mediated decay pathway. EMBO J 26: 1820–1830 [Europe PMC free article] [Abstract] [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J (2018) fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 34: i884–i890 [Europe PMC free article] [Abstract] [Google Scholar]
- Cheng Z, Muhlrad D, Lim MK, Parker R, Song H (2007) Structural and functional insights into the human Upf1 helicase core. EMBO J 26: 253–264 [Europe PMC free article] [Abstract] [Google Scholar]
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, Mühlemann O (2017) Transcriptome‐wide identification of NMD‐targeted human mRNAs reveals extensive redundancy between SMG6‐ and SMG7‐mediated degradation pathways. RNA 23: 189–201 [Europe PMC free article] [Abstract] [Google Scholar]
- Costa‐Mattioli M, Walter P (2020) The integrated stress response: From mechanism to disease. Science 368: eaat5314 [Europe PMC free article] [Abstract] [Google Scholar]
- Czaplinski K, Weng Y, Hagan KW, Peltz SW (1995) Purification and characterization of the Upf1 protein: a factor involved in translation and mRNA degradation. RNA 1: 610–623 [Europe PMC free article] [Abstract] [Google Scholar]
- Dockendorff TC, Labrador M (2019) The fragile X protein and genome function. Mol Neurobiol 56: 711–721 [Abstract] [Google Scholar]
- Durand S, Franks TM, Lykke‐Andersen J (2016) Hyperphosphorylation amplifies UPF1 activity to resolve stalls in nonsense‐mediated mRNA decay. Nat Commun 7: 1–12 [Europe PMC free article] [Abstract] [Google Scholar]
- Eberle AB, Lykke‐Andersen S, Mühlemann O, Jensen TH (2009) SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16: 49–55 [Abstract] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 1–7 [Europe PMC free article] [Abstract] [Google Scholar]
- Fiorini F, Bonneau F, Le Hir H (2012) Biochemical characterization of the RNA helicase UPF1 involved in nonsense‐mediated mRNA decay. Methods Enzymol 511: 255–274 [Abstract] [Google Scholar]
- Fiorini F, Boudvillain M, Le Hir H (2013) Tight intramolecular regulation of the human Upf1 helicase by its N‐ and C‐terminal domains. Nucleic Acids Res 41: 2404–2415 [Europe PMC free article] [Abstract] [Google Scholar]
- Franks TM, Singh G, Lykke‐Andersen J (2010) Upf1 ATPase‐dependent mRNP disassembly is required for completion of nonsense‐ mediated mRNA decay. Cell 143: 938–950 [Europe PMC free article] [Abstract] [Google Scholar]
- Fritz SE, Haque N, Hogg JR (2018) Highly efficient in vitro translation of authentic affinity‐purified messenger ribonucleoprotein complexes. RNA 24: 982–989 [Europe PMC free article] [Abstract] [Google Scholar]
- Fritz SE, Ranganathan S, Wang CD, Hogg JR (2020) The RNA‐binding protein PTBP1 promotes ATPase‐dependent dissociation of the RNA helicase UPF1 to protect transcripts from nonsense‐mediated mRNA decay. J Biol Chem 295: 11613–11625 [Europe PMC free article] [Abstract] [Google Scholar]
- Gautier A, Juillerat A, Heinis C, Corrêa IR Jr, Kindermann M, Beaufils F, Johnsson K (2008) An engineered protein tag for multiprotein labeling in living cells. Chem Biol 15: 128–136 [Abstract] [Google Scholar]
- Ge Z, Quek BL, Beemon KL, Hogg JR (2016) Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense‐mediated mRNA decay pathway. eLife 5: e11155 [Europe PMC free article] [Abstract] [Google Scholar]
- Ge Z, Quek BL, Beemon KL, Hogg JR (2016) Gene Expression Omnibus GSE59884 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59884). [DATASET]
- Gehring NH, Kunz JB, Neu‐Yilik G, Breit S, Viegas MH, Hentze MW, Kulozik AE (2005) Exon‐junction complex components specify distinct routes of nonsense‐mediated mRNA decay with differential cofactor requirements. Mol Cell 20: 65–75 [Abstract] [Google Scholar]
- Goetz AE, Wilkinson M (2017) Stress and the nonsense‐mediated RNA decay pathway. Cell Mol Life Sci 74: 3509–3531 [Europe PMC free article] [Abstract] [Google Scholar]
- Goldstein JL, Brown MS (2009) The LDL receptor. Arterioscler Thromb Vasc Biol 29: 431–438 [Europe PMC free article] [Abstract] [Google Scholar]
- Gowravaram M, Bonneau F, Kanaan J, Maciej VD, Fiorini F, Raj S, Croquette V, Le Hir H, Chakrabarti S (2018) A conserved structural element in the RNA helicase UPF1 regulates its catalytic activity in an isoform‐specific manner. Nucleic Acids Res 46: 2648–2659 [Europe PMC free article] [Abstract] [Google Scholar]
- Harbeck N, Penault‐Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J, Cardoso F (2019) Breast cancer. Nat Rev Dis Primers 5: 66 [Abstract] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [Europe PMC free article] [Abstract] [Google Scholar]
- Hodgkin J, Papp A, Pulak R, Ambros V, Anderson P (1989) A new kind of informational suppression in the nematode Caenorhabditis elegans . Genetics 123: 301–313 [Europe PMC free article] [Abstract] [Google Scholar]
- Hogg JR, Collins K (2007a) RNA‐based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA 13: 868–880 [Europe PMC free article] [Abstract] [Google Scholar]
- Hogg JR, Collins K (2007b) Human Y5 RNA specializes a Ro ribonucleoprotein for 5S ribosomal RNA quality control. Genes Dev 21: 3067–3072 [Europe PMC free article] [Abstract] [Google Scholar]
- Hogg JR, Goff SP (2010) Upf1 senses 3′ UTR length to potentiate mRNA decay. Cell 143: 379–389 [Europe PMC free article] [Abstract] [Google Scholar]
- Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum‐localized mRNAs during the unfolded protein response. Science 313: 104–107 [Abstract] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009a) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [Abstract] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009b) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [Europe PMC free article] [Abstract] [Google Scholar]
- Huang L, Lou C‐H, Chan W, Shum EY, Shao A, Stone E, Karam R, Song H‐W, Wilkinson MF (2011) RNA homeostasis governed by cell type‐specific and branched feedback loops acting on NMD. Mol Cell 43: 950–961 [Europe PMC free article] [Abstract] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Saulière J, Izaurralde E (2008) SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14: 2609–2617 [Europe PMC free article] [Abstract] [Google Scholar]
- Hurt JA, Robertson AD, Burge CB (2013) Global analyses of UPF1 binding and function reveal expanded scope of nonsense‐mediated mRNA decay. Genome Res 23: 1636–1650 [Europe PMC free article] [Abstract] [Google Scholar]
- Jan CH, Williams CC, Weissman JS (2014) Principles of ER cotranslational translocation revealed by proximity‐specific ribosome profiling. Science 346: 1257521 [Europe PMC free article] [Abstract] [Google Scholar]
- Karousis ED, Mühlemann O (2019) Nonsense‐mediated mRNA decay begins where translation ends. Cold Spring Harb Perspect Biol 11: a032862 [Europe PMC free article] [Abstract] [Google Scholar]
- Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S (2006) Binding of a novel SMG‐1‐Upf1‐eRF1‐eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense‐mediated mRNA decay. Genes Dev 20: 355–367 [Europe PMC free article] [Abstract] [Google Scholar]
- Kearse MG, Goldman DH, Choi J, Nwaezeapu C, Liang D, Green KM, Goldstrohm AC, Todd PK, Green R, Wilusz JE (2019) Ribosome queuing enables non‐AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev 33: 871–885 [Europe PMC free article] [Abstract] [Google Scholar]
- Kearse M, Goldman D, Choi J, Nwaezeapu C, Liang D, Green K, Goldstrohm A, Todd P, Green R, Wilusz J (2019) Gene Expression Omnibus GSE125086 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125086). [DATASET]
- Keene JD (2007) RNA regulons: coordination of post‐transcriptional events. Nat Rev Genet 8: 533–543 [Abstract] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [Europe PMC free article] [Abstract] [Google Scholar]
- Kim YK, Maquat LE (2019) UPFront and center in RNA decay: UPF1 in nonsense‐mediated mRNA decay and beyond. RNA 25: 407–422 [Europe PMC free article] [Abstract] [Google Scholar]
- Kishor A, Fritz SE, Hogg JR (2019a) Nonsense‐mediated mRNA decay: the challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip Rev RNA 10: e1548 [Europe PMC free article] [Abstract] [Google Scholar]
- Kishor A, Fritz SE, Haque N, Ge Z, Tunc I, Yang W, Zhu J, Hogg JR (2020) Activation and inhibition of nonsense‐mediated mRNA decay control the abundance of alternative polyadenylation products. Nucleic Acids Res 48: 7468–7482 [Europe PMC free article] [Abstract] [Google Scholar]
- Kishor A, Ge Z, Hogg JR (2019b) hnRNP L‐dependent protection of normal mRNAs from NMD subverts quality control in B cell lymphoma. EMBO J 38: e99128 [Europe PMC free article] [Abstract] [Google Scholar]
- Kurosaki T, Li W, Hoque M, Popp MW‐L, Ermolenko DN, Tian B, Maquat LE (2014) A post‐translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev 28: 1900–1916 [Europe PMC free article] [Abstract] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE (2007) Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446: 926–929 [Abstract] [Google Scholar]
- Lavysh D, Neu‐Yilik G (2020) UPF1‐Mediated RNA decay‐danse macabre in a cloud. Biomolecules 10: 999 [Europe PMC free article] [Abstract] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ (2000a) The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon‐exon junctions. EMBO J 19: 6860–6869 [Europe PMC free article] [Abstract] [Google Scholar]
- Le Hir H, Moore MJ, Maquat LE (2000b) Pre‐mRNA splicing alters mRNP composition: evidence for stable association of proteins at exon‐exon junctions. Genes Dev 14: 1098–1108 [Europe PMC free article] [Abstract] [Google Scholar]
- Lee SR, Pratt GA, Martinez FJ, Yeo GW, Lykke‐Andersen J (2015) Target discrimination in nonsense‐mediated mRNA decay requires Upf1 ATPase activity. Mol Cell 59: 413–425 [Europe PMC free article] [Abstract] [Google Scholar]
- Li Z, Vuong JK, Zhang M, Stork C, Zheng S (2017) Inhibition of nonsense‐mediated RNA decay by ER stress. RNA 23: 378–394 [Europe PMC free article] [Abstract] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [Abstract] [Google Scholar]
- Linares AJ, Lin C‐H, Damianov A, Adams KL, Novitch BG, Black DL (2015) The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. eLife 4: e09268 [Europe PMC free article] [Abstract] [Google Scholar]
- Linares AJ, Lin CH, Damianov A, Adams KL, Novitch BG, Black D (2015) Gene Expression Omnibus GSE71179 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71179). [DATASET]
- Loh B, Jonas S, Izaurralde E (2013) The SMG5‐SMG7 heterodimer directly recruits the CCR4‐NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev 27: 2125–2138 [Europe PMC free article] [Abstract] [Google Scholar]
- Longman D, Jackson‐Jones KA, Maslon MM, Murphy LC, Young RS, Stoddart JJ, Hug N, Taylor MS, Papadopoulos DK, Cáceres JF (2020) Identification of a localized nonsense‐mediated decay pathway at the endoplasmic reticulum. Genes Dev 34: 1075–1088 [Europe PMC free article] [Abstract] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 10–12 [Google Scholar]
- Martinez‐Nunez RT, Sanford JR (2016) Gene Expression Omnibus GSE89774 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89774). [DATASET]
- Martinez‐Nunez RT, Wallace A, Coyne D, Jansson L, Rush M, Ennajdaoui H, Katzman S, Bailey J, Deinhardt K, Sanchez‐Elsner T et al (2017) Modulation of nonsense mediated decay by rapamycin. Nucleic Acids Res 45: 3448–3459 [Europe PMC free article] [Abstract] [Google Scholar]
- Mathews MB, Hershey JWB (2015) The translation factor eIF5A and human cancer. Biochim Biophys Acta 1849: 836–844 [Europe PMC free article] [Abstract] [Google Scholar]
- Michlewski G, Cáceres JF (2019) Post‐transcriptional control of miRNA biogenesis. RNA 25: 1–16 [Europe PMC free article] [Abstract] [Google Scholar]
- de Miguel FJ, Sharma RD, Pajares MJ, Montuenga LM, Rubio A, Pio R (2014) Identification of alternative splicing events regulated by the oncogenic factor SRSF1 in lung cancer. Cancer Res 74: 1105–1115 [Abstract] [Google Scholar]
- Mugridge JS, Coller J, Gross JD (2018) Structural and molecular mechanisms for the control of eukaryotic 5’‐3' mRNA decay. Nat Struct Mol Biol 25: 1077–1085 [Abstract] [Google Scholar]
- Nathans D (1964) Puromycin inhibition of protein synthesis: incorporation of puromycin into peptide chains. Proc Natl Acad Sci USA 51: 585–592 [Europe PMC free article] [Abstract] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr (2007) Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense‐mediated decay. Genes Dev 21: 708–718 [Europe PMC free article] [Abstract] [Google Scholar]
- Nicholson P, Josi C, Kurosawa H, Yamashita A, Mühlemann O (2014) A novel phosphorylation‐independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res 42: 9217–9235 [Europe PMC free article] [Abstract] [Google Scholar]
- Nickless A, Jackson E, Marasa J, Nugent P, Mercer RW, Piwnica‐Worms D, You Z (2014) Intracellular calcium regulates nonsense‐mediated mRNA decay. Nat Med 20: 961–966 [Europe PMC free article] [Abstract] [Google Scholar]
- Niknafs YS, Pandian B, Iyer HK, Chinnaiyan AM, Iyer MK (2017) TACO produces robust multisample transcriptome assemblies from RNA‐seq. Nat Methods 14: 68–70 [Europe PMC free article] [Abstract] [Google Scholar]
- Page MF, Carr B, Anders KR, Grimson A, Anderson P (1999) SMG‐2 is a phosphorylated protein required for mRNA surveillance in Caenorhabditis elegans and related to Upf1p of yeast. Mol Cell Biol 19: 5943–5951 [Europe PMC free article] [Abstract] [Google Scholar]
- Park Y, Reyna‐Neyra A, Philippe L, Thoreen CC (2017) mTORC1 balances cellular amino acid supply with demand for protein synthesis through post‐transcriptional control of ATF4. Cell Rep 19: 1083–1090 [Europe PMC free article] [Abstract] [Google Scholar]
- Park Y, Reyna‐Neyra A, Philippe L, Thoreen CC (2017) Gene Expression Omnibus GSE97384 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97384). [DATASET]
- Paz I, Kosti I, Ares M Jr, Cline M, Mandel‐Gutfreund Y (2014) RBPmap: a web server for mapping binding sites of RNA‐binding proteins. Nucleic Acids Res 42: W361–W367 [Europe PMC free article] [Abstract] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T‐C, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat Biotechnol 33: 290–295 [Europe PMC free article] [Abstract] [Google Scholar]
- Powell DR (2015) Degust: interactive RNA‐seq analysis. 10.5281/zenodo.3258933 [CrossRef]
- Pulak R, Anderson P (1993) mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev 7: 1885–1897 [Abstract] [Google Scholar]
- Risso D, Ngai J, Speed TP, Dudoit S (2014) Normalization of RNA‐seq data using factor analysis of control genes or samples. Nat Biotechnol 32: 896–902 [Europe PMC free article] [Abstract] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [Europe PMC free article] [Abstract] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2009) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [Europe PMC free article] [Abstract] [Google Scholar]
- Schofield JA, Duffy EE, Kiefer L, Sullivan MC, Simon MD (2018) TimeLapse‐seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat Methods 15: 221–225 [Europe PMC free article] [Abstract] [Google Scholar]
- Shen S, Park JW, Lu Z‐X, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA‐Seq data. Proc Natl Acad Sci USA 111: E5593–E5601 [Europe PMC free article] [Abstract] [Google Scholar]
- Singh G, Rebbapragada I, Lykke‐Andersen J (2008) A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense‐mediated mRNA decay. PLoS Biol 6: e111 [Europe PMC free article] [Abstract] [Google Scholar]
- Smith JE, Baker KE (2015) Nonsense‐mediated RNA decay–a switch and dial for regulating gene expression. BioEssays 37: 612–623 [Europe PMC free article] [Abstract] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13: 283–296 [Abstract] [Google Scholar]
- The Gene Ontology Consortium (2019) The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47: D330–D338 [Europe PMC free article] [Abstract] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [Europe PMC free article] [Abstract] [Google Scholar]
- Toma KG, Rebbapragada I, Durand S, Lykke‐Andersen J (2015) Identification of elements in human long 3’ UTRs that inhibit nonsense‐mediated decay. RNA 21: 887–897 [Europe PMC free article] [Abstract] [Google Scholar]
- UniProt Consortium (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49: D480–D489 [Europe PMC free article] [Abstract] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J‐Y, Cody NAL, Dominguez D et al (2020) A large‐scale binding and functional map of human RNA‐binding proteins. Nature 583: 711–719 [Europe PMC free article] [Abstract] [Google Scholar]
- Vitting‐Seerup K, Sandelin A (2019) IsoformSwitchAnalyzeR: analysis of changes in genome‐wide patterns of alternative splicing and its functional consequences. Bioinformatics 35: 4469–4471 [Abstract] [Google Scholar]
- Waldron JA, Tack DC, Ritchey LE, Gillen SL, Wilczynska A, Turro E, Bevilacqua PC, Assmann SM, Bushell M, Le Quesne J (2019) Gene Expression Omnibus. GSE134888 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134888). [DATASET]
- Waldron JA, Tack DC, Ritchey LE, Gillen SL, Wilczynska A, Turro E, Bevilacqua PC, Assmann SM, Bushell M, Le Quesne J (2019) mRNA structural elements immediately upstream of the start codon dictate dependence upon eIF4A helicase activity. Genome Biol 20: 1–23 [Europe PMC free article] [Abstract] [Google Scholar]
- Watson M, Park Y, Thoreen C (2021) Roadblock‐qPCR: A simple and inexpensive strategy for targeted measurements of mRNA stability. RNA 27: 335–342 [Europe PMC free article] [Abstract] [Google Scholar]
- Wek RC (2018) Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb Perspect Biol 10: a032870 [Europe PMC free article] [Abstract] [Google Scholar]
- Weng Y, Czaplinski K, Peltz SW (1998) ATP is a cofactor of the Upf1 protein that modulates its translation termination and RNA binding activities. RNA 4: 205–214 [Europe PMC free article] [Abstract] [Google Scholar]
- Yee BA, Pratt GA, Graveley BR, Van Nostrand EL, Yeo GW (2019) RBP‐Maps enables robust generation of splicing regulatory maps. RNA 25: 193–204 [Europe PMC free article] [Abstract] [Google Scholar]
- Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Mühlemann O (2011) Autoregulation of the nonsense‐mediated mRNA decay pathway in human cells. RNA 17: 2108–2118 [Europe PMC free article] [Abstract] [Google Scholar]
- Yi Z, Sanjeev M, Singh G (2021) The Branched Nature of the Nonsense‐Mediated mRNA Decay Pathway. Trends Genet 37: 143–159 [Europe PMC free article] [Abstract] [Google Scholar]
- Young SK, Wek RC (2016) Upstream open reading frames differentially regulate gene‐specific translation in the integrated stress response. J Biol Chem 291: 16927–16935 [Europe PMC free article] [Abstract] [Google Scholar]
- Young‐Baird SK, Lourenço MB, Elder MK, Klann E, Liebau S, Dever TE (2020) Suppression of MEHMO syndrome mutation in eIF2 by small molecule ISRIB. Mol Cell 77: 875–886 [Europe PMC free article] [Abstract] [Google Scholar]
- Zünd D, Gruber AR, Zavolan M, Mühlemann O (2013) Translation‐dependent displacement of UPF1 from coding sequences causes its enrichment in 3’ UTRs. Nat Struct Mol Biol 20: 936–943 [Abstract] [Google Scholar]
Reviewer Report
Articles from The EMBO Journal are provided here courtesy of Nature Publishing Group
Full text links
Read article at publisher's site: https://doi.org/10.15252/embj.2021108898
Read article for free, from open access legal sources, via Unpaywall:
https://onlinelibrary.wiley.com/doi/pdfdirect/10.15252/embj.2021108898

Citations & impact
Impact metrics
Citations of article over time
Alternative metrics

Discover the attention surrounding your research
https://www.altmetric.com/details/126429289
Smart citations by scite.ai
Explore citation contexts and check if this article has been
supported or disputed.
https://scite.ai/reports/10.15252/embj.2021108898
Article citations
Nonsense-Mediated mRNA Decay in Human Health and Diseases: Current Understanding, Regulatory Mechanisms and Future Perspectives.
Mol Biotechnol, 12 Sep 2024
Cited by: 0 articles | PMID: 39264527
Review
UPF1 helicase orchestrates mutually exclusive interactions with the SMG6 endonuclease and UPF2.
Nucleic Acids Res, 52(10):6036-6048, 01 Jun 2024
Cited by: 1 article | PMID: 38709891
junctionCounts: comprehensive alternative splicing analysis and prediction of isoform-level impacts to the coding sequence.
NAR Genom Bioinform, 6(3):lqae093, 09 Aug 2024
Cited by: 0 articles | PMID: 39131822 | PMCID: PMC11310779
UPF1 ATPase autoinhibition and activation modulate RNA binding kinetics and NMD efficiency.
Nucleic Acids Res, 52(9):5376-5391, 01 May 2024
Cited by: 0 articles | PMID: 38412299 | PMCID: PMC11109973
Messenger RNA Surveillance: Current Understanding, Regulatory Mechanisms, and Future Implications.
Mol Biotechnol, 27 Feb 2024
Cited by: 1 article | PMID: 38411790
Review
Go to all (15) article citations
Data
Data behind the article
This data has been text mined from the article, or deposited into data resources.
BioStudies: supplemental material and supporting data
Ensembl Genome Browser (2)
- (3 citations) Ensembl - ENST00000599848
- (2 citations) Ensembl - ENST00000262803
Protein structures in PDBe (2)
-
(1 citation)
PDBe - 6EJ5View structure
-
(1 citation)
PDBe - 2XZPView structure
Similar Articles
To arrive at the top five similar articles we use a word-weighted algorithm to compare words from the Title and Abstract of each citation.
The RNA-binding protein PTBP1 promotes ATPase-dependent dissociation of the RNA helicase UPF1 to protect transcripts from nonsense-mediated mRNA decay.
J Biol Chem, 295(33):11613-11625, 22 Jun 2020
Cited by: 18 articles | PMID: 32571872 | PMCID: PMC7450115
Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway.
Elife, 5:e11155, 08 Jan 2016
Cited by: 86 articles | PMID: 26744779 | PMCID: PMC4764554
Activation and inhibition of nonsense-mediated mRNA decay control the abundance of alternative polyadenylation products.
Nucleic Acids Res, 48(13):7468-7482, 01 Jul 2020
Cited by: 11 articles | PMID: 32542372 | PMCID: PMC7367170
UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond.
RNA, 25(4):407-422, 17 Jan 2019
Cited by: 110 articles | PMID: 30655309 | PMCID: PMC6426291
Review
 Free full text in Europe PMC
Free full text in Europe PMC
Funding
Funders who supported this work.


